


 下载:
下载:

-
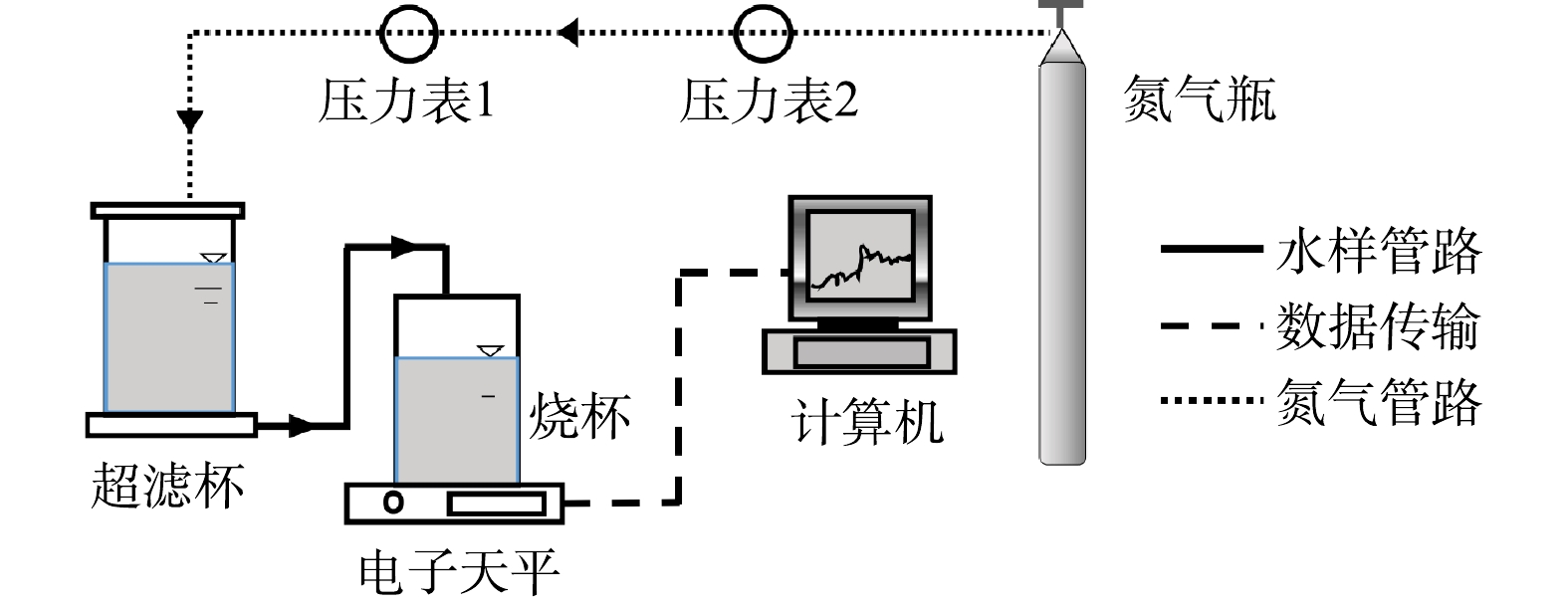
将混凝作为超滤(ultrafiltration,UF)预处理工艺,通过调控膜前过滤水中颗粒形态及共存污染物的赋存状态,优化沉积于膜表面滤饼层的结构和性质,从而可有效提高膜通量、改善膜滤效果和减缓膜污染[1-3]。与传统化学絮凝不同,加载絮凝(ballasted flocculation,BF)工艺利用高分子助凝剂水解产物的架桥作用,促使加入混凝剂后形成的微絮体与所投加的载体颗粒(通常选用微砂)相结合,不断成长为加载絮体,可有效改善已形成凝聚体的沉降性能及抗剪切能力,并减轻进水负荷波动的影响,提高净水效能[4]。特别地,由于微砂的表面电荷密度较低,可强化凝聚体对水中重金属、有机污染物等的吸附性能,使得这些污染物主要以悬浮态形式存在于经加载絮凝处理的水中[5]。对于加载絮凝-超滤(BF-UF)净水过程,助凝剂投加量决定着高分子链上可提供的有效吸附位点的数量[6-9],而原水pH则关系着混凝剂和助凝剂的水解产物及表面电荷[10-12],均会对加载絮体形态以及后续超滤阶段的滤饼层结构和膜污染程度产生显著影响。基于此,探究助凝剂投加量及原水pH对加载絮凝与超滤联用时膜污染行为的影响特性具有重要意义。
本研究以高岭土、腐殖酸和Sb3+的混合液为模拟原水,聚丙烯酰胺(polyacrylamide,PAM)为助凝剂,在分别考察PAM投加量、原水pH对超滤阶段膜通量衰减和膜污染可逆性等影响的基础上,通过分析加载絮体形态特征及其与膜污染的相关性,并结合宏观出水水质变化,进一步探讨了各考察条件对BF-UF组合工艺膜污染的影响机制,以期为超滤组合工艺的优选提供参考。
全文HTML
-
模拟水样:以高岭土(SiO2 46%,Al2O3 39%,分析纯)、腐殖酸(HA,化学纯)和酒石酸锑钾(KSbOC4H4O6·1/2H2O ≥99%,分析纯)分别作为原水中浊质颗粒、天然有机物和重金属污染物的代表,与超纯水配制成实验水样,其主要水质参数:浊度为(100±1) NTU、UV254为(0.200±0.005) cm−1、Sb3+ 为50 μg·L−1。
化学试剂:混凝剂采用聚硫酸铁(PFS,相对密度为1.45,分析纯),助凝剂为聚丙烯酰胺(PAM,阳离子型,分子质量为8 000~15 000 kDa,分析纯),用超纯水分别配制成质量浓度为5.0 g·L−1(以PFS计)和1.0 g·L−1(以PAM计)的溶液后直接投加;盐酸和氢氧化钠(均为分析纯)用以调整模拟水样的pH;载体颗粒选用粒径为75~106 μm的微砂。超滤膜使用聚偏氟乙烯(PVDF)平板超滤膜,切割分子质量为100 kDa。
-
采用混凝六联搅拌仪(TA6-2,武汉恒岭)开展加载絮凝实验,依照GHANEM等[13]提出的小试方案设置搅拌强度(G)及时间,即首先向水样中投加混凝剂PFS,并以536 r·min−1(G1 = 600 s−1)快速搅拌1 min,而后投加助凝剂PAM,继续快搅10 s,接下来加入微砂颗粒,以相同的转速再搅拌10 s,最后以258 r·min−1(G2 = 200 s−1)搅拌2 min完成加载絮体成长。开展单因素加载絮凝实验时,助凝剂PAM按0、0.2、0.3、0.5、0.6、1.0 mg·L−1投加,模拟水样pH分别调至4、5、6、7、8、9。除被考察因素之外,其他均设定为预先确定的最佳实验条件:PFS为30 mg·L−1,PAM为0.3 mg·L−1,微砂为3 g·L−1,pH为7。
超滤实验装置及系统如图1所示。将上述不同PAM投加量和pH条件下经加载絮凝处理后的水样转移至装有PVDF平板膜的超滤杯(MSC300,上海摩速)中,并由氮气提供驱动力(保持恒压0.10 MPa),出水质量用与电子天平(NVL511B,上海奥豪斯)相连的计算机进行实时记录,计算恒压膜通量(JV,其中V表示过滤絮凝水样的累加体积)。每个超滤过程分为3个过滤周期,各周期依次过滤200 mL超纯水和240 mL加载絮凝水样,并在每1个周期结束后均将膜片从超滤杯中取出,先借助振荡器在固定的振幅和频率下振荡3 min,再对膜片正反面进行水力冲洗,以去除膜面污染物。
-
加载絮体微观特征:采用凝聚体平均粒径和分形维数来表征加载絮体形态,二者提取及计算方法同文献中的方法[14];借助电位分析仪(ZS90,英国马尔文)分别测定絮凝体系中投加PFS和PAM后以及絮体成熟阶段的Zeta电位值。
膜污染评价:超滤过程中膜污染程度用膜比通量(JV/J0)随过滤絮凝水样体积V的变化趋势来反映;表征膜污染可逆性的指标包括可逆污染值(R可逆)、不可逆污染值(R不可逆)和总污染值(R总),均由膜通量按式(1)~式(3)求得[2]。
式中:J0为新膜的纯水通量,L·(m2·h)−1;Je为过滤结束时的膜通量,L·(m2·h)−1;Jw为受污染膜清洗后的纯水通量,L·(m2·h)−1。
宏观出水水质分别采用浊度仪(SGZ-200AS,上海悦丰)、紫外可见分光光度计(U-3900,日本日立)、原子荧光光度计(AFS-9700,北京海光)测定加载絮凝后水样、超滤膜滤出水的浊度、UV254和Sb3+含量,用以辅助膜污染机理的探讨。
1.1. 实验材料
1.2. 加载絮凝-超滤实验
1.3. 分析方法
-
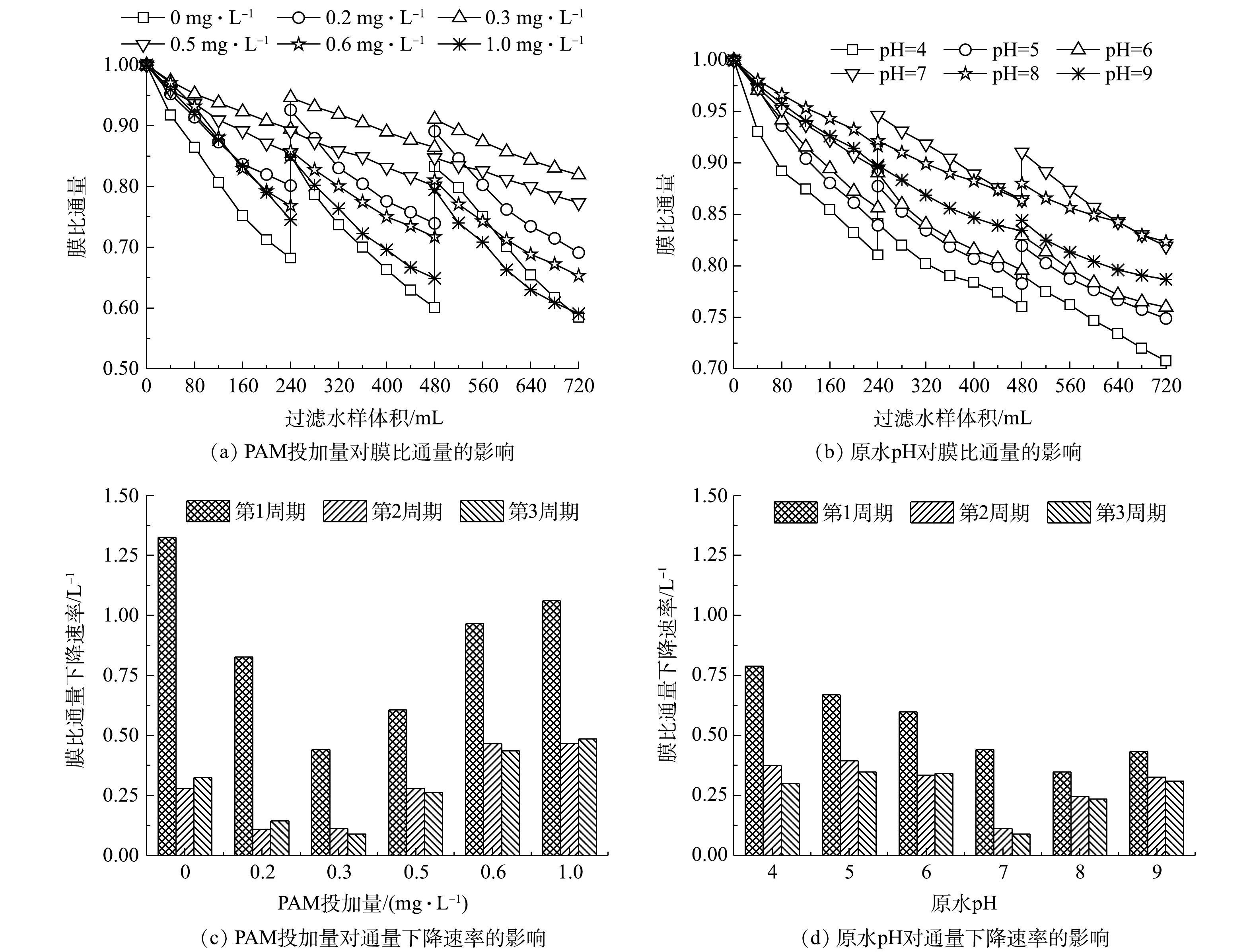
1)PAM投加量及pH对各超滤周期内膜比通量变化的影响特征。助凝剂(PAM)投加量及原水pH条件下加载絮凝-超滤组合工艺的膜污染程度,可用膜比通量随过滤絮凝水样体积的变化来表示,结果如图2所示。在任一PAM投加量对应的超滤过程中,各周期内膜比通量均呈下降趋势,并且经过水洗后下一周期膜比通量的初始值和末端值也低于前一周期的相应值(图2(a));与此同时,第1周期的膜比通量下降速率(即同一周期内膜比通量下降量与过滤水样体积之比)均远大于第2和第3周期(图2(c))。类似地,仅改变原水pH时,也可观察到相同的膜比通量变化趋势(图2(b)和图2(d))。由此可见,随着过滤的进行,由吸附和堵塞等引起的累积不可逆膜污染[10]会逐渐加剧。
2) PAM投加量对加载絮凝-超滤组合工艺膜滤性能及膜污染的影响特征。由图2(a)并结合图2(c)可看出,加载絮凝阶段中的PAM投加量对超滤过程中各周期的末端膜比通量及膜比通量下降速率均有显著影响。在PAM投加量由0 mg·L−1增至0.3 mg·L−1的过程中,同一周期的末端膜比通量逐渐增大,分别由0.682 1增至0.894 3(第1周期)、由0.600 7增至0.864 4(第2周期)、由0.584 6增至0.819 1(第3周期),而膜比通量下降速率骤降,其中第1、2和3周期的相应值分别减小了66.74%、59.61%、72.81%;此后,随着助凝剂量的继续增加,2个特征值的变化趋势与0~0.3 mg·L−1内的刚好相反,即同一周期的膜比通量下降速率开始加快,致使各周期的末端膜比通量均不断减小。
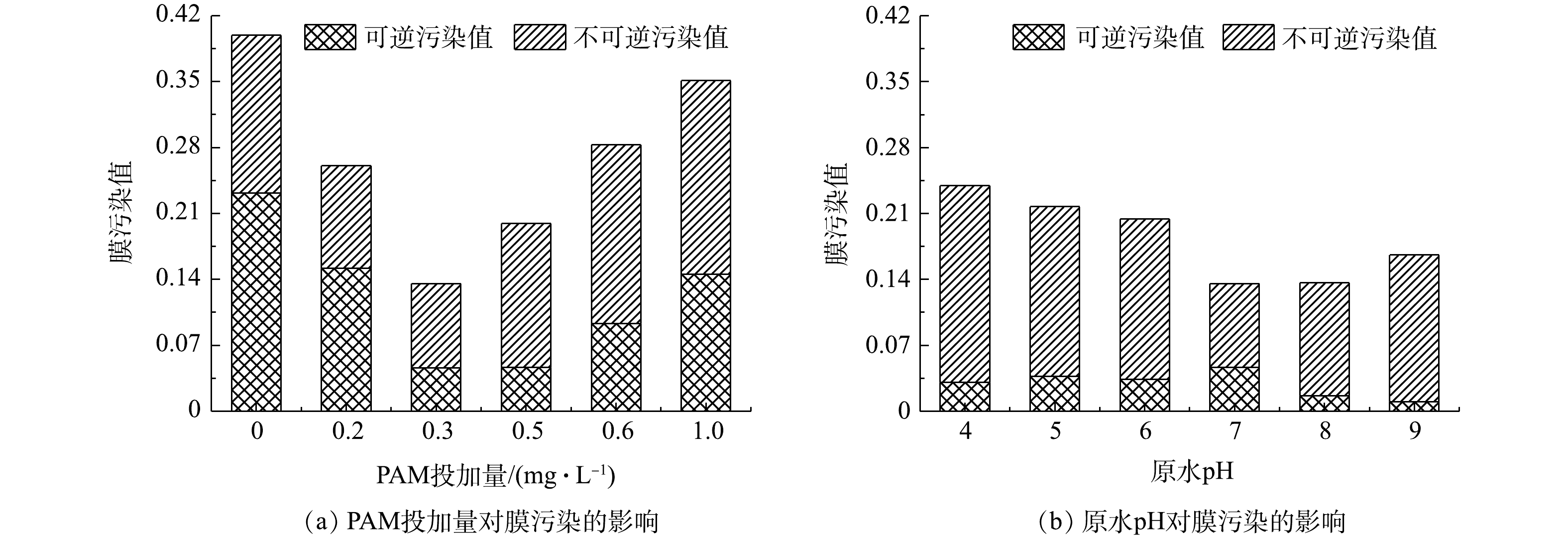
为进一步探究PAM投加量对超滤膜污染的影响,图3(a)给出了由式(1)~式(3)计算得出的运行3个周期之后的膜污染值。当助凝剂投加量在0~0.3 mg·L−1内增加时,不可逆污染值和总污染值各下降了46.82%和66.05%,并在0.3 mg·L−1时二者均取得最小值;当继续增大PAM投加量至1.0 mg·L−1,上述2个污染指标将比PAM投加量为0.3 mg·L−1时分别增加了1.30倍和1.59倍。显然,只有向加载絮凝体系中投加适量的PAM(0.3 mg·L−1),才能有效削弱超滤阶段膜不可逆污染和总污染的发生,而若助凝剂的投加量不足或过多,均不利于提高超滤膜的抗污染能力。其原因是,不同PAM投加量下膜表面形成滤饼层的结构差异明显,对膜过滤性能和膜污染程度会产生较大的影响[15]。
3)原水pH对加载絮凝-超滤组合工艺膜滤性能及膜污染的影响特征。在pH由4增加至7的过程中,第1、2和3周期的末端膜比通量分别由0.810 7、0.760 3、0.707 5提高到0.894 3、0.864 4、0.819 1,变化幅度各为10.31%、13.70%、15.78%(图2(b)),而同一周期内膜比通量下降速率依次减少了44.16%、70.04%、70.60%(图2(d))。这表明,相较于酸性溶液环境,中性条件更有利于维持较高的膜比通量以及显著减缓膜比通量的下降速率,此时膜污染程度最轻。上述结论同样可由图3(b)得出,即原水pH由4增至7时,不可逆污染值和总污染值分别由0.208 9减小至0.089 3、由0.239 7减小至0.135 6,降幅高达57.28%和43.45%,这说明膜不可逆污染和总污染均得到有效的抑制。在偏碱性条件(pH为8和9)下,尽管各周期的末端膜比通量大多数略高于pH为7时的同一周期值(图2(b)),但在给定周期内膜比通量下降速率随着pH的增大呈现加快趋势(图2(d)),并且较大的pH对应的膜不可逆污染值和总污染值均较高(图3(b))。从膜通量变化和膜污染可逆性的角度综合考虑,中性条件优于偏碱性条件,而酸性条件的结果最不理想,这与童少磊等[16]在探究pH对传统化学絮凝-超滤组合工艺的膜污染影响时所得到的的结论基本一致。
-
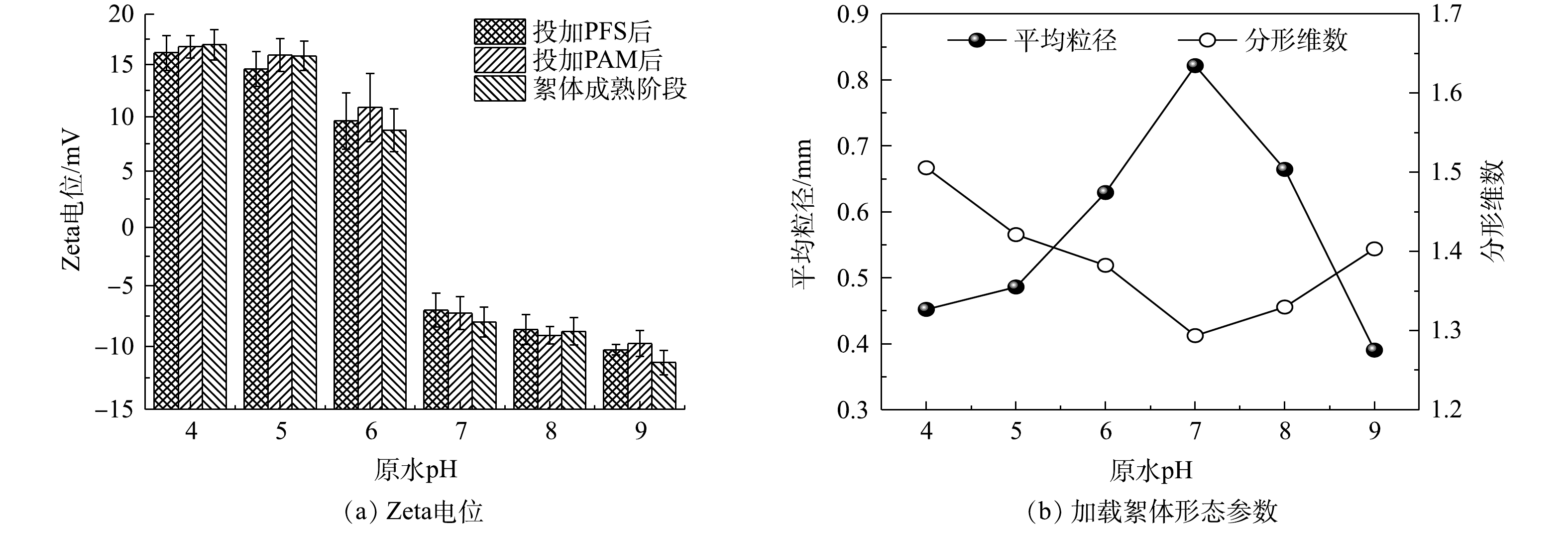
就混凝与超滤组合工艺而言,诸多研究者[1, 3, 17]认为,膜前待滤液内絮体的带电特性及形态特征决定着超滤膜上累积形成滤饼层的结构,继而对膜过滤效能和膜污染程度产生影响。基于此,本节首先考察PAM投加量、原水pH分别对加载絮凝体系的Zeta电位变化以及所形成絮体的平均粒径和分形维数等形态参数的影响规律,结果如图4和图5所示。
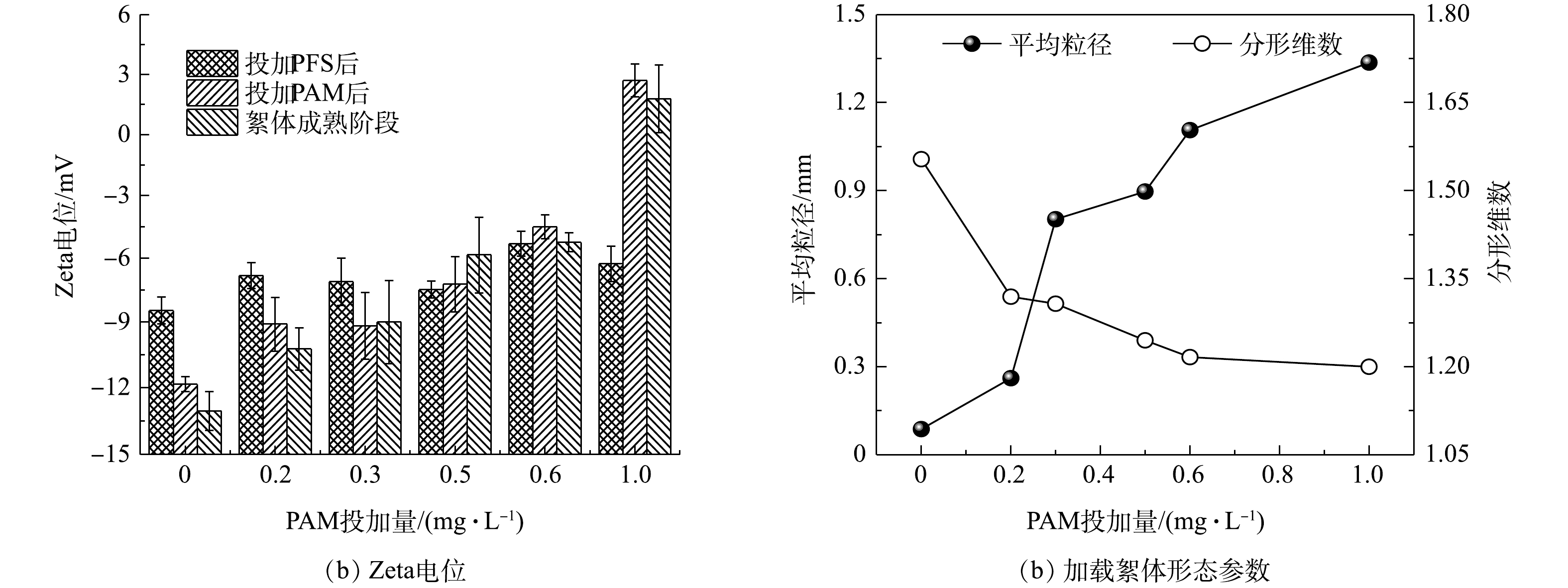
由图4(a)可知,向pH为7的模拟原水中投加30 mg·L−1 PFS后,Zeta电位虽然比投加混凝剂前的(−21.93±0.87) mV(注:图中未给出)更接近于0 mV,但所形成的微絮体仍呈电负性(对应于图中“投加PFS后”的Zeta电位),极易与PAM高分子链上的阳离子基团发生静电吸附作用[18],促使加载絮凝体系中已形成微絮体与具有较低表面电荷密度的微砂颗粒在阳离子型PAM的架桥作用下不断成长为粒径较大的加载絮体。结合图4(b)所示的PAM投加量对絮体形态参数的影响进一步分析可得,未投加助凝剂或投加量较小(如0.2 mg·L−1)时,有机高分子链上吸附位点数量严重不足,导致其对微絮体和微砂颗粒的捕获能力(架桥作用)非常弱,难以形成大絮体(平均粒径最大值仅为0.26 mm);当PAM投量增至0.3~0.5 mg·L−1时,水中显著增多的吸附位点有利于生成更多大粒径范围的絮体,此时平均粒径高达0.85 mm左右;但若继续增加投量(0.6 mg·L−1和1.0 mg·L−1),会诱使絮体疯狂成长,且结构开始变得更为疏松。其原因可能是,在PFS、PAM和微砂三者投加比例不协调的情形下,过量的高分子助凝剂在促使成熟阶段加载絮体快速成长的同时,高分子水解产物上的活性基团与高岭土颗粒之间还会产生专性吸附,进而加速了不含微砂絮体的大量形成,导致加载絮体的占比大幅减小[8-9, 19]。
相较于PAM投加量,原水pH对加载絮凝阶段不同时刻Zeta电位的影响更为明显(图5(a))。在酸性条件(pH = 4~6)下,投加混凝剂PFS后的Zeta电位由模拟水样的(−15.27±0.76) mV(图中未给出)迅速转为正值,即形成了带正电荷的微絮体,在加入0.3 mg·L−1 PAM后,体系Zeta电位继续保持正值,但其大小随着pH的增加逐渐减小;在中性及偏碱性条件下,模拟水样的Zeta电位在(−22.44±0.75) mV附近波动,在投加PFS、PAM后以及絮体成熟阶段的Zeta电位仍均为负值,并且其绝对值随着pH的增加有所增大。由图5(b)可知,原水pH在4~9逐渐增加时,加载絮体的平均粒径呈先增大而后骤减的趋势,分形维数的变化规律与之相反,且二形态参数均在pH为7时达到极值,此时生成大而结构松散的加载絮体。产生上述现象的原因主要是由于:原水pH决定着混凝剂PFS和助凝剂PAM的水解产物及表面电荷,进而对加载絮体成长产生显著影响[12, 16, 18],即pH在4~7时逐渐增加时,PFS的水解产物
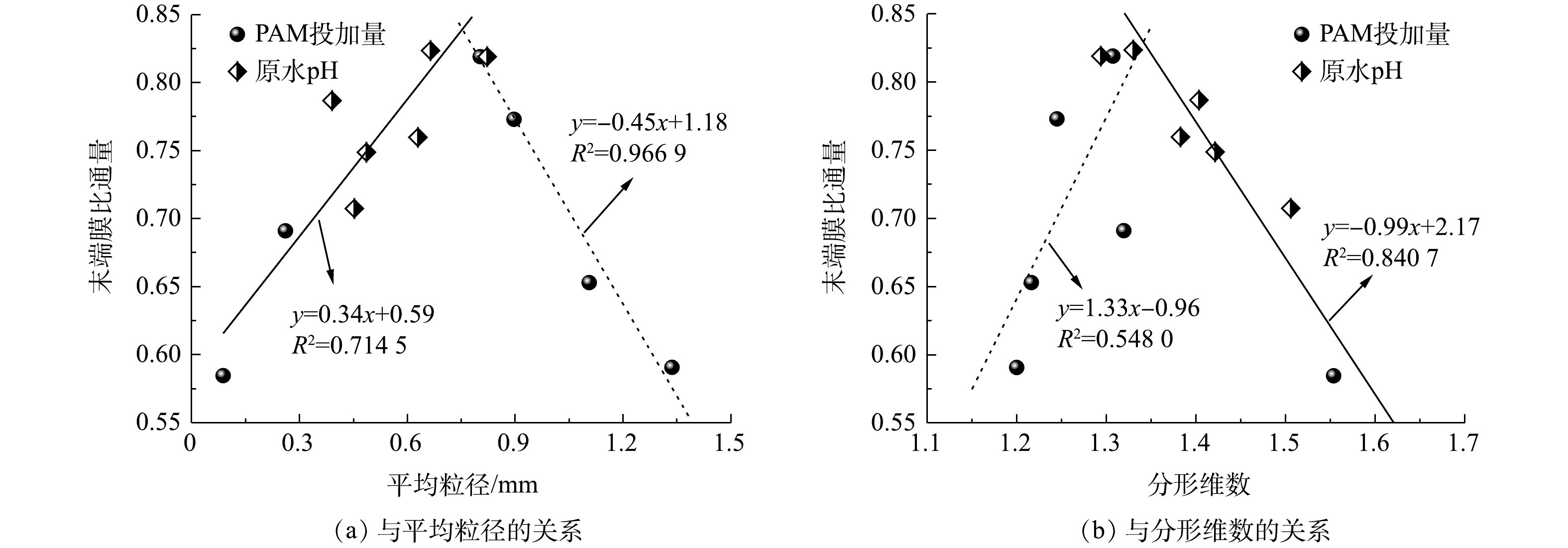
$ {\rm{Fe}}\left( {{\rm{OH}}} \right)_2^ + $ 等正价态聚合离子迅速增多,且此时PAM高分子链处于稳定状态并保持高度伸展,有利于后者架桥作用的发挥;但若pH>7,PFS的水解产物将以无定型的Fe(OH)3和带负电荷的$ {\rm{Fe}}\left( {{\rm{OH}}} \right)_4^ - $ 为主要存在形式,并且PAM在此条件下因不断解离出羧基而导致其阳离子电荷密度下降,同时其长链延伸程度也受到影响,有效吸附位点数量大幅降低,致使加载絮凝过程的吸附电中和与架桥作用均被削弱,进而阻碍絮体的长大。为了进一步解析加载絮体形态对超滤膜污染的影响效应,现将第3周期末端膜比通量分别与加载絮体平均粒径、分形维数进行线性拟合,结果如图6所示。有研究[17]表明,絮体粒径越大、结构越疏松(对应着较小的分形维数),超滤过程中形成滤饼层的空间结构越有利于缓解膜污染,这一基于传统化学絮凝-超滤组合工艺构建的相关关系仅在加载絮体平均粒径小于0.75 mm、分形维数大于1.35时成立,拟合度R2分别为0.714 5(图6(a))和0.840 7(图6(b))。然而,对于平均粒径大于0.75 mm、分形维数小于1.35的加载絮体,末端膜比通量与平均粒径之间却存在着显著的负相关性(R2 = 0.966 9)(图6(a)),并且随着分形维数的减小,末端膜比通量呈下降趋势(图6(b)),表明此时由粒径较大、结构较为松散的凝聚体在膜表面形成的滤饼层并未能有效缓解膜污染,对应着较小的末端膜比通量。结合图4(b)和图5(b)可见,平均粒径大于0.75 mm以及分形维数小于1.35的加载絮体主要是在PAM投加量为0.3~1.0 mg·L−1(仅改变助凝剂量)条件下形成的,即较高的助凝剂量是导致加载絮体过度生长、末端膜比通量下降的主要原因。这可能是由于絮体在膜表面堆积而成的滤饼层因结构特征、可压缩性和抗污染能力等方面各异导致的[10](原因解析详见2.3节)。
-
如上文所述,在加载絮凝阶段所形成絮体的特性,会影响超滤膜表面滤饼层结构及水中溶解态污染物的存在形态,进而关系着超滤阶段膜过滤效能和膜污染行为。图7和图8分别给出了PAM投加量和原水pH对加载絮凝-超滤组合工艺膜污染生成以及各运行阶段的浊质颗粒、腐殖酸和Sb3+等污染物去除效果的影响情况。
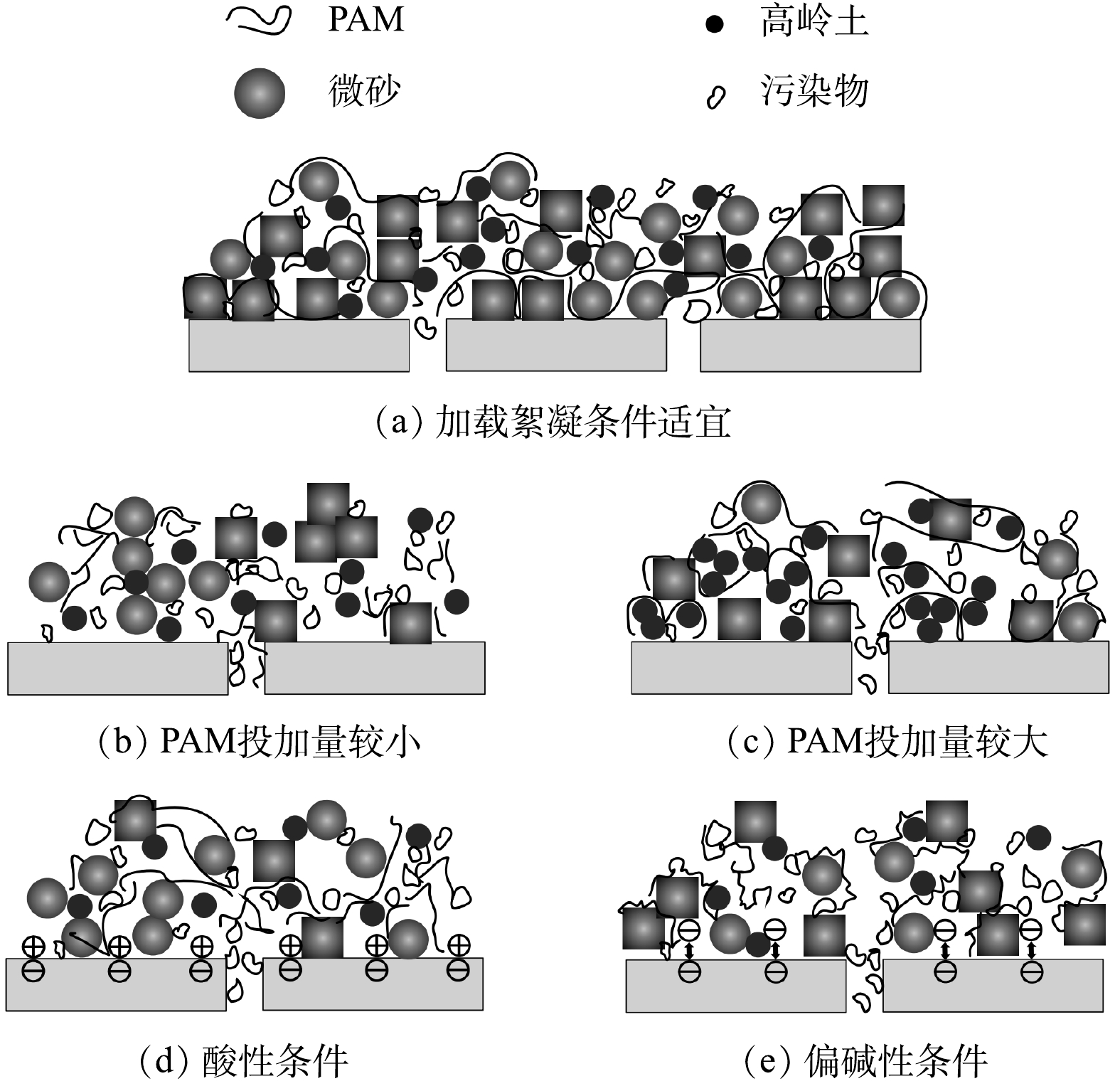
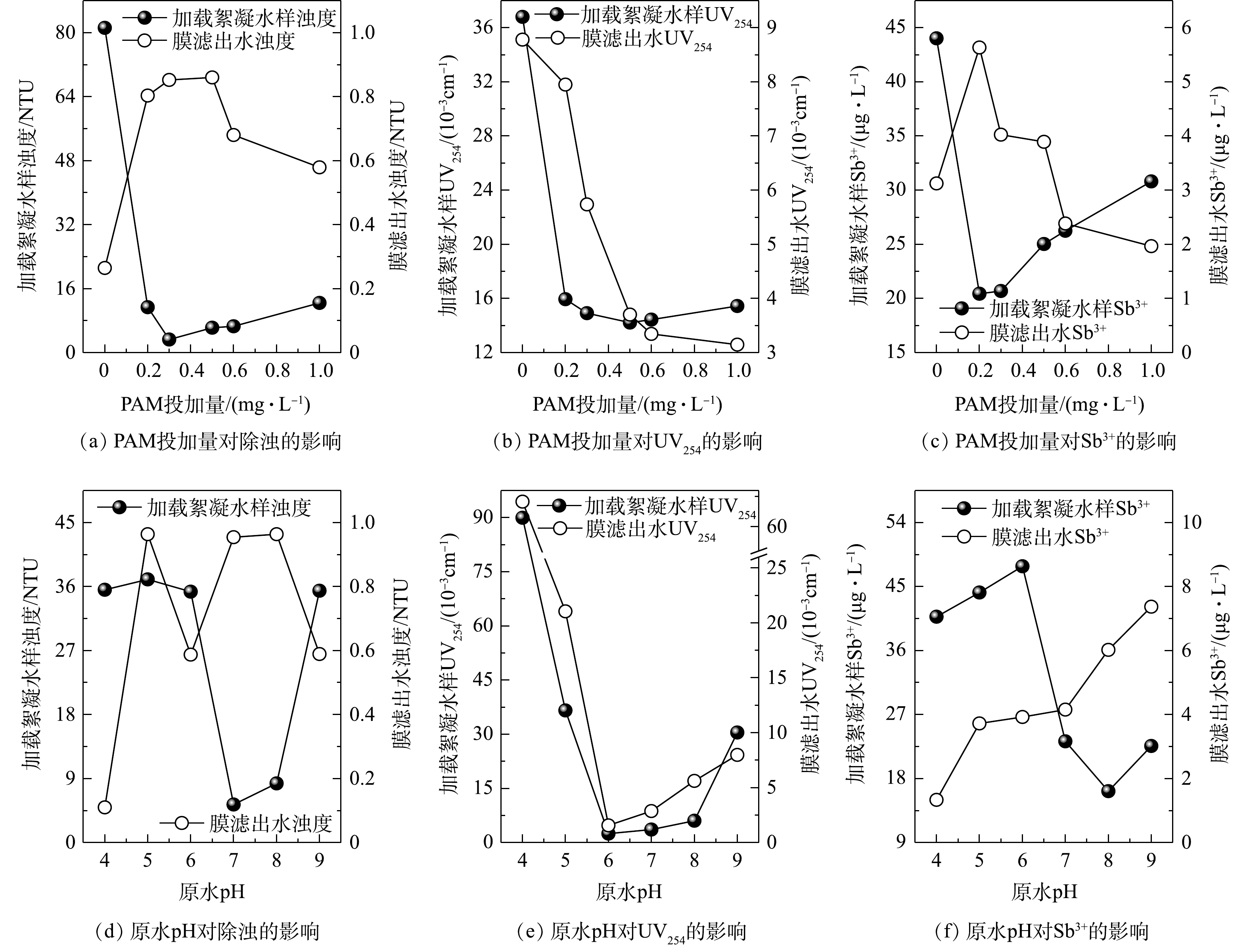
由图8可看出,在PAM投加量为0.3 mg·L−1和原水pH为7的条件下,经加载絮凝处理后水样的浊度、UV254和Sb3+浓度均处于较低水平。这表明加载絮凝阶段形成絮体的粒径较大且结构较为疏松(图4(b)和图5(b)),有利于强化原水中腐殖酸和Sb3+等各类污染物吸附于絮体表面而呈悬浮态[5]。结合图2和图3可知:待滤液中小粒径颗粒以及溶解态污染物的大幅减少,可有效避免他们在超滤初期直接与膜材料接触,有助于控制因膜孔吸附和堵塞等引起的不可逆膜污染;随着超滤过程的进行,加载絮体会在膜表面不断累积形成一层具有特殊结构的疏松多孔的滤饼层(图7(a)),通过滤饼层的吸附与筛分过滤的协同作用可进一步延缓膜污染(尤其是不可逆污染)的发生和发展,并达到较好的超滤净水效果(图8)。其可能原因主要有2点:一是进入滤饼层的微砂颗粒,因其表面电荷密度较低且粒径远大于超滤膜孔径,会再次强化吸附膜滤液中溶解态有机物和重金属离子等污染物,并在最大程度上将其拦截于滤饼层内,从而在稳定超滤净水水质的同时,可继续减少各类污染物与膜表面接触的机会,有效降低膜表面的污染负荷;二是微砂颗粒还可被视作滤饼层的骨架,能够最大化地提高后者的抗压缩变形能力和孔隙率,以便超滤净水过程中维持滤饼层内部过滤通道的畅通,不至于造成膜比通量下降速率的显著加快。
超滤膜表面的滤饼层结构与加载絮体形态特征密切相关,而助凝剂投加量决定着加载絮凝体系中高分子链上可提供有效吸附位点的数量,继而对最终形成絮体的大小和松散程度等产生较大影响[9-10, 18]。对于未投加助凝剂或PAM投加量较小(0.2 mg·L−1)的工况而言,加入高分子助凝剂后用于微絮体和微砂颗粒架桥连接的有效吸附位点数量较少,致使絮凝末期形成絮体的粒径非常小且结构较为致密(图4(b)),同时还有可能造成部分微砂颗粒因无法获得有效吸附位点而始终游离于加载絮凝水样中,均不利于加载絮凝阶段各类污染物的吸附去除。在2种工况下,浊度、UV254和Sb3+浓度分别为PAM投加量为0.3 mg·L−1时的24.81倍和3.47倍(图8(a))、2.47倍和1.07倍(图8(b))、2.13倍和0.99倍(图8(c))。此后,将对含有大量的溶解态有机物和小粒度絮体的加载絮凝水样进行超滤处理,在过滤初期极有可能因堵塞膜孔而引起较大的不可逆膜污染和总污染(图3(a)),并且过滤中后期由小絮体及微砂颗粒在膜表面形成滤饼层的结构较为致密(图7(b)),导致其透水性变差,对应着较小的末端膜比通量和较大的通量下降速率(图2(a)和图2(c))。
当PAM投加量增至0.5~1.0 mg·L−1时,加载絮凝阶段水中显著增多的吸附位点有助于形成粒径更大且结构更为松散的絮体(图4(b)),使得经加载絮凝处理后待滤液中可能引起膜不可逆污染的溶解态有机物含量维持在较低水平,对应的UV254值与PAM投加量为0.3 mg·L−1时的差别不大(图8(b)),但在超滤净水过程中的膜比通量下降速率及膜污染值均高于后者的相应值(图2(c)和图3(a)),并且根据2.2节的分析可知,在上述投加量下末端膜比通量与加载絮体各形态参数之间并未表现出基于传统化学絮凝-超滤组合工艺构建的相关关系(图6)。其原因是,在PFS、PAM和微砂三者投加比例不协调的情形下,过量的高分子助凝剂虽然可为加载絮凝体系中微砂颗粒与已形成微絮体的架桥连接提供足够的吸附位点,促使成熟阶段加载絮体快速成长,但因PAM水解产物上的活性基团与高岭土颗粒之间存在着专性吸附作用[19],同样加速了不含微砂絮体的大量形成,并且随着PAM投加量的增大,通过专性吸附形成絮体的占比可能更大,于是超滤阶段形成滤饼层内微砂颗粒的空间分布极不均匀,其骨架作用并未得到有效发挥(图7(c)),致使超滤净水过程中滤饼层的抗压缩能力不足,极易变得密实,从而加剧了膜污染。
结合图5的结果可知,不同原水pH下形成的加载絮体在带电特性、粒径和结构等方面同样存在较大差异。其中,在酸性条件(pH=4~6)下加入PFS后形成的微絮体带正电,与阳离子型PAM之间的静电排斥作用可使后者高分子链处于高度伸展的稳定状态[7],并且随着pH的增大,混凝剂和助凝剂的水解产物更有利于絮凝末期形成粒径大、结构松散的加载絮体,对溶解态有机物的吸附去除效果提升明显,表现为相较于pH为4和6时经加载絮凝处理后水样的UV254值降幅高达97.26%(图8(e))。但原水pH由4增至6的过程中,运行3个过滤周期之后膜总污染值、不可逆污染值的差别并不大(图3(b)),即待滤液中溶解态有机物的不断减少以及超滤阶段膜表面形成更为疏松多孔的滤饼层(图7(d)),并未能有效抑制膜污染(尤其是不可逆污染)的发生。这可能是因为,带正电的加载絮体在膜表面累积形成的滤饼层与呈负电性的PVDF超滤膜之间的静电吸引作用,既加剧了膜表面的污染负荷,又增大了采用简单水洗来恢复膜通量的难度[10]。
在偏碱性条件(pH>7)下,投加PFS后致使加载絮凝体系中形成的微絮体带负电,继而引起阳离子型PAM的高分子链收缩卷曲[7]。pH越大,投加PAM前已形成微絮体的负电性越强(图5(a)),使得高分子长链延伸程度受到的影响进一步增大,越不利于形成粒径大且结构松散的加载絮体(图5(b)),最终在超滤膜表面形成更为致密的滤饼层(图7(e));与此同时,经加载絮凝处理后水样(膜前液)的浊度、UV254和Sb3+浓度也随之增大(图8(d)和图8(e))),均会造成膜污染加重(图3(b))。此外,根据形成滤饼层的加载絮体与PVDF超滤膜均呈电负性,可推知滤饼层与膜之间的静电排斥作用能够在一定程度上阻碍污染物附着于膜表面,同时可改善污染物从膜表面上被水洗去除的效果。这即为前文提及的偏碱性条件下膜不可逆污染值和总污染值均比酸性条件下的相应值低的可能原因之一。
2.1. PAM投加量及pH对超滤膜污染的影响
2.2. 加载絮体形态与膜污染的相关性


2.3. 膜污染影响机制分析
-
1)助凝剂PAM的投加量不足或较大时,均不利于超滤净水过程中膜污染(尤其是不可逆污染)的缓解,对应着较小的末端膜比通量和较大的通量下降速率。这主要是由于,助凝剂投加量关系着加载絮凝体系中可提供有效吸附位点的数量,继而对加载絮体成长及滤饼层形成产生较大影响。
2)在酸性条件下,随着pH的增大,由带正电的加载絮体在膜表面累积形成的滤饼层更为疏松多孔,并且待滤液中溶解态有机物也减少,但滤饼层与PVDF超滤膜之间的静电作用致使膜表面的污染负荷仍高于中性条件下;在偏碱性条件下,滤饼层与膜之间静电排斥效应阻碍了污染物粘附于膜表面,对缓解膜污染起到积极作用。
3)基于传统化学絮凝-超滤组合工艺构建的线性关系仅在加载絮体平均粒径小于0.75 mm、分形维数大于1.35时成立。
4)在PAM投加量为0.3 mg·L−1和原水pH为7的条件下,可最大化地减少加载絮凝水样中溶解态污染物的含量以及游离态的微砂颗粒和不含微砂的凝聚体等的占比,随后由加载絮体在膜表面累积形成的滤饼层能够充分发挥吸附和筛分过滤的协同作用,从而达到延缓膜污染和稳定超滤出水水质的目的。
