

-
土壤作为承载和积累环境污染物的一个“汇”,其污染问题不可小觑,因此,污染土壤的治理和修复研究具有重要意义。在众多修复技术中,电动修复(electrokinetic remediation, EK)技术由于不会对土壤造成扰动、修复周期短、可处理低渗透性土壤等优势受到研究者们的关注,其基本原理是在土壤两端施加一定的电压梯度,利用电迁移、电渗流和电泳等电动力学现象将土壤中的污染物运输到两端的电极区,再作进一步的处理[1]。对于有机污染土壤,常采用原位投加化学氧化剂并通过电化学驱动其到土体中降解有机污染物的方式进行修复,称为电动-原位化学氧化技术(electrokinetic in situ chemical oxidation, EK-ISCO)[2-4]。
EK-ISCO技术在处理过程中由于污染物降解和阴极还原会导致氧化剂的消耗[5-6],因此需定期外源投加氧化剂,同时氧化剂(例如双氧水、过硫酸盐)的购买、运输和保存等也会增加成本[7]。此外,由于传统的电动修复研究中仅关注电极附近水解产生的H+和OH−对土壤pH的影响[8-9],而忽视了阳极处发生的氧化反应对土壤有机污染物的降解作用,且大量电能因在阳极处发生析氧反应而浪费。因此研究者们提出利用阳极原位自产氧化剂而无需外源投加氧化剂,并可通过电动迁移的方式输送到土体(阳极产生的自由基寿命短,无法通过溶液传输),从而实现有机污染土壤的修复。SONG等[10]较早开展了相关研究,采用RuO2/Ti阳极在NaCl电解质中自产活性氯来去除石英砂土柱中的石油烃污染物,土柱孔隙水中活性氯的最高浓度达到30 mmol·L−1,获得最高67.1%的石油烃去除率。目前该研究刚刚起步,对于阳极产生的氧化剂如何在土柱中输送以及对污染物的去除机制仍不甚清楚。
在阳极自产氧化剂过程中,电极材料和反应条件是影响氧化剂产生效率的主要影响因素。不同电极材料的研究较多,包括掺硼金刚石(boron-doped diamond,BDD)电极、形稳电极(dimensionally stabel anodes,DSA,一般为钛基多金属电极)和铂电极等[11-12]。近年来研究发现Ti4O7电极的电导率高,耐强酸强碱,且析氧电位较高,是一种性能优良的新型电极材料[13]。一般而言,电极的析氧电位越高,则产生氧化剂的能力越强,此外也与电解液的组分有关。GRGUR等[14]在使用RuO2/Ti电极降解水体中的多灭威时加入了氯化钠(浓度为2 g·L−1),结果表明,在电流密度为10-20 mA cm−2时0.5 h即可降解90%以上的多灭威,其中活性氯的产率最高达0.018 mmol·min−1。FARHAT等[15]选择BDD电极,研究了在不同电解质溶液中(硫酸钠、高氯酸钠、硝酸钠等)自产氧化剂的可行性,结果表明,BDD电极在硫酸钠溶液中能够产生过硫酸盐氧化剂;基本反应过程有2个:一是SO42-在阳极表面直接失去电子形成SO4·−(式(1));另一种方式是水在阳极表面形成·OH,·OH与SO42-反应生成SO4·−(式(2)~(3)),上述反应生成的SO4·−可两两聚合形成 S2O82−(式(4))。然而,上述研究未能对不同电极自产氧化剂的条件和能力进行比较,难以对修复实践提供指导。
本研究选择BDD、Ti4O7和RuO2/Ti 3类典型的阳极材料作为阳极,以硫酸钠溶液为电解质,以常见的土壤中多环芳烃蒽作为典型有机污染物,考察了不同电极在不同条件下产生过硫酸盐氧化剂的能力,阐明阳极自产过硫酸盐在土柱中的迁移规律和对污染物的去除,以期为EK-ISCO技术的发展提供新的技术支持。
-
本研究选用的阳极材料共4种,均为方形片状电极,分别为2种BDD电极,1种Ti4O7电极,1种RuO2/Ti电极(表层镀氧化钌的钛电极)。电极厚度均为1 mm,其中2种BDD电极购自湖南新峰科技有限公司,尺寸分别为8 cm×4 cm和4 cm×4 cm;Ti4O7电极尺寸为4 cm×4 cm,购自安徽正影科技有限公司;RuO2/Ti尺寸为4 cm×4 cm,购自苏州舒尔泰工业科技有限公司。
石英砂 (分析纯) 购自中国医药集团有限公司,使用前将石英砂用1 mol·L−1硫酸浸泡1 d,去离子水冲洗至中性,65 ℃烘6 h,保存备用。阳离子交换膜(cation exchange membrane,CEM)为CMI-7000S型,购自美国(Membranes International INC., New Jersey)。蒽和其他化学试剂均为分析纯,购自中国医药集团有限公司,所有试剂均用去离子水进行配制。
-
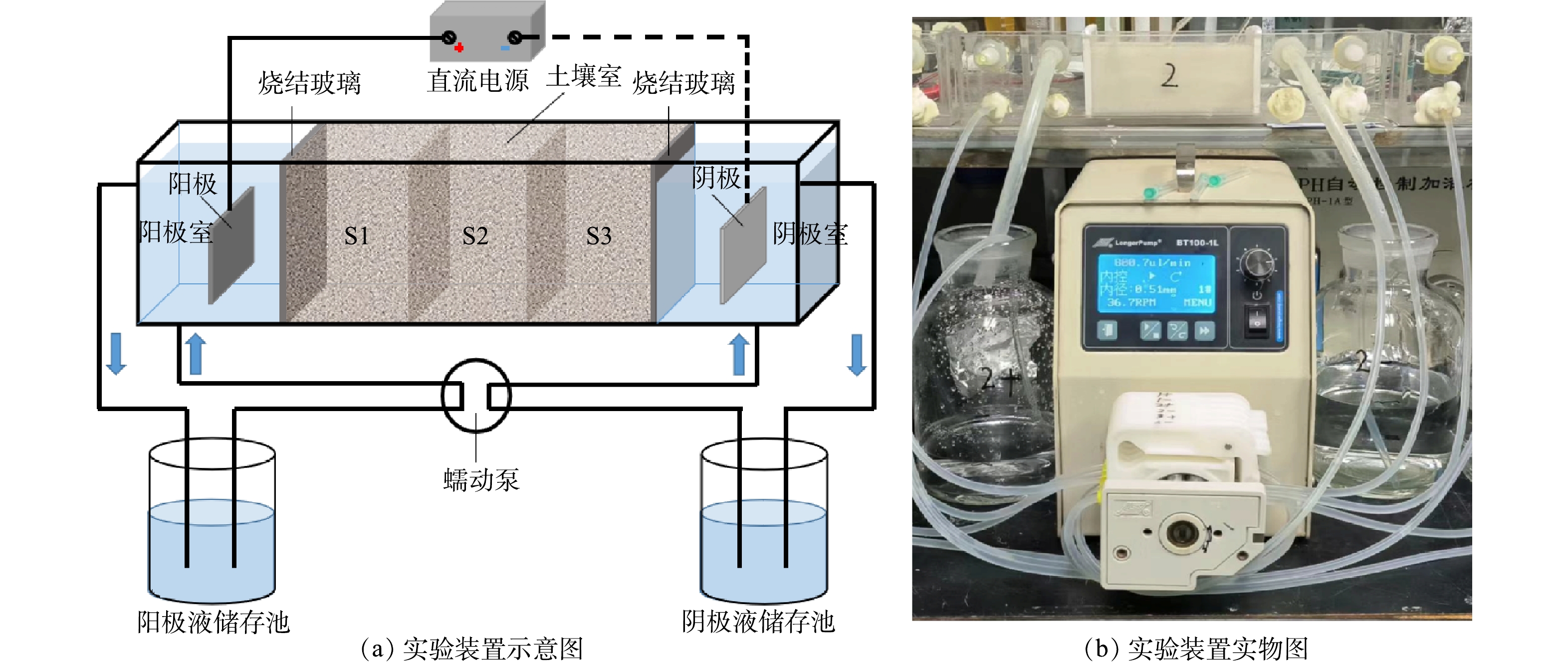
实验装置采用图1所示的长方体反应池,用有机玻璃制成,内部尺寸为长6 cm×宽4 cm×高12 cm。以选用的4种电极材料为阳极,以4 cm×4 cm RuO2/Ti为阴极,施加一定电流密度的直流电(直流电源为DP832型,30 V/3 A,普源精电科技股份有限公司,中国),开展不同条件下的电极自产氧化剂实验(表1),研究阳极自产氧化剂的主要影响因素。反应池中的硫酸钠溶液体积为200 mL,实验开始时用浓硫酸调节溶液pH为2[15]。
-
实验采用图2的土柱实验装置。实验装置为长30 cm、宽8 cm、高6 cm的长方形有机玻璃盒,分为土壤室(12 cm×8 cm×6 cm)、阴极室(8.5 cm×8 cm×6 cm)和阳极室(8.5 cm×8 cm×6 cm),土壤室与电极室之间通过多孔烧结玻璃隔开以避免土壤室中的固体颗粒进入电极室。电极室通过蠕动泵与电极液储槽相连并不断循环电极液,流速为10 mL·min−1。在实验开始前,在土壤室中加入700 g 洗净后的石英砂,将其均匀分为3个部分,从阳极到阴极分别记为S1、S2和S3,并于每个截面中心位置预埋入3个土壤溶液采集器[16],用0.01 mol·L−1硝酸钠溶液饱和2 d。
阳极采用尺寸均为4 cm×4 cm的BDD(T1)、Ti4O7(T2)、RuO2/Ti(T3)3种电极,使用RuO2/Ti(4 cm×4 cm)作为阴极。采用0.6 mol·L−1的硫酸钠溶液作为阳极液,0.01 mol·L−1的硝酸钠作为阴极液。在两极间施加20 V直流电压(根据常规电动修复的电压设置)[10,17],通电时间为6 h。在实验过程中,分别在0、2、6 h记录电流和电渗流,并采集阴、阳极电极液和土柱中3个截面的土壤溶液,测定溶液的pH、EC、PS浓度和自由基等。
-
采用的实验装置同1.3节,其中土壤室装填的石英砂改为人为配制的蒽(anthracene, ANT)污染石英砂,装填量为700 g。蒽污染石英砂的配制方法为:在洗净石英砂中添加定量溶有ANT的丙酮溶液(固液比1:1.5,即100 g土加150 mL丙酮),通风橱内避光挥发24 h,石英砂中ANT最终含量为49.5 mg·kg−1。装柱时预埋入土壤溶液采集器,定期采集土壤溶液。实验设计4个处理,为T4~T7,采用0.6 mol·L−1的硫酸钠溶液作为阳极液,0.01 mol·L−1的硝酸钠作为阴极液。T4~T6采用3种不同阳极材料,分别为BDD电极(T4)、Ti4O7电极(T5)、RuO2/Ti(T6),尺寸均为4 cm×4 cm,以RuO2/Ti作为阴极,电压为20 V,通电时间为24 h。同时设置T7作为对照处理,即不使用电极,其他条件和T4~T6一致,研究在不通电的情况下石英砂中ANT的自然降解情况。在实验过程中,分别在0、2、6、12和24 h记录电流和电渗流,并采集土壤溶液和电解液样品,测定溶液的pH、EC、PS浓度等。实验结束时,采集不同土柱截面的石英砂,冻干保存,测定ANT浓度。
-
实验过程中电流由直流电源(IT6322A,艾德克斯电子有限公司,中国)监测;电渗流通过定期记录阳极液体积变化得出,以阳极液体积的减少量为电渗流量;溶液样品的pH和EC分别用pH计(S210-K,梅特勒托利多仪器有限公司,瑞士)和电导率仪(FE38,梅特勒托利多仪器有限公司,瑞士)测定;溶液中PS的浓度采用紫外分光光度法测定,即在试管中依次加入 3.9 mL 去离子水和1 mL 250 g·L−1 KI 和 12.5 g·L−1 NaHCO3混合溶液,再加入0.1 mL待测溶液,显色后用紫外可见分光光度计(M4型,上海美谱达仪器有限公司,中国)在 400 nm 处测定(检测限0.01 mmol·L−1)。
阴阳极溶液和土壤溶液中的自由基分析采用电子顺磁共振波谱仪(Bruker E500-9.5/12,布鲁克仪器有限公司, 德国)测定。具体方法为抽取阴阳极溶液和土壤溶液,立即过0.45 μm滤膜,迅速加入10 μL DMPO溶液,混合均匀制备成待测溶液,送电子顺磁共振波谱仪测定。用毛细管吸取待测溶液并封口,放置于电子自旋共振仪中测定待测溶液中的·OH和SO4·−[18]。石英砂中ANT的浓度测定参照DONG等[19]的方法,采用二氯甲烷-正己烷超声萃取,通过GC-MS(GCMS-QP2010 Ultra,岛津,日本)测定。
-
图3为不同影响因素对阳极自产氧化剂的影响。如图3(a)所示,电流密度同为15 mA·cm−1的情况下,用CEM膜将阳极室和阴极室分隔开,120 min后PS的最高浓度从1.54 mmol·L−1提高到3.44 mmol·L−1,表明CEM可防止过硫酸根扩散到阴极,抑制了PS在阴极的分解。YUKSELEN-AKSOY等[20]和LI等[21]的研究发现,过硫酸盐在阴极会与多余电子和还原性H2发生还原反应分解生成SO42-,导致PS大量损失。XU等[8]将CEM应用于EK-ISCO中处理有机污染土壤时,也证实CEM可以防止PS接触阴极,减少损失,从而提高污染物的去除效率。本研究中,用CEM将阳极室和阴极室隔离开,此时阳极室测定的PS含量能更准确地代表阳极自产PS 的能力,因此,在后续实验中均使用CEM分隔阴、阳极室,以便更准确地分析阳极自产氧化剂的真实性能。
图3(b)反映了不同电流密度下阳极自产过硫酸盐浓度的变化。在较低的电流密度下,电极表面电荷转移受到限制,导致PS的生成速率较慢,因此,当电流密度由7.5 mA cm−2分别增加到15 mA·cm−2和30 mA·cm−2时,120 min内PS浓度由1.44 mmol·L−1增加到3.44 mmol·L−1和8.78 mmol·L−1,这也与FARHAT等的研究结果一致[15]。电解质浓度是硫酸盐转化为PS的基础。如图3(c)所示,当Na2SO4浓度由0.05 mol·L−1增加到0.6 mol·L−1时,产生的PS浓度随Na2SO4浓度增加而增加,在Na2SO4浓度为0.6 mol·L−1时,PS浓度达到最大(105.1 mmol·L−1)。但当Na2SO4浓度继续增加时,PS浓度反而开始下降。这是因为阳极表面的吸附位点是有限的,当过多的SO42-迁移到电极表面被吸附时,阻碍了·OH的生成,从而抑制电化学氧化生成PS的速率[22]。另一种可能的原因是溶液中过量的SO4−·发生自消耗反应,致使溶液中检测到的PS浓度减少(式(5))[23-24]。
通电时间对PS浓度的影响结果见图3(d)。随着通电时间的延长,PS的浓度逐步升高,1 000 min时达到25 mmol·L−1,此后随着时间的延长,PS的浓度缓慢增加。这表明在2 000 min内,电催化生成PS的过程是可以持续进行的,这可为之后进行较长时间的阳极自产PS降解土柱中有机污染物的研究奠定基础。
电极是电化学氧化系统的核心,与氧化剂的电化学产生和污染物降解效率直接相关。阳极上的析氧反应与·OH生成反应为竞争反应,因此,较高的析氧电位有助于抑制析氧反应,促进·OH的生成[25],并提高能源利用效率。由图3(e)可见,不同阳极自产PS的量有明显不同。在电极面积相同时,BDD电极处理中PS浓度最高,120 min时达到10 mmol·L−1,其次是Ti4O7电极,为2 mmol·L−1,最后是RuO2/Ti,仅为0.50 mmol·L−1。这与3种电极的析氧电位关系密切,一般来说析氧电位越高,则产生PS的能力越强。3种电极中BDD电极的析氧电位最高,达2.80 V,而Ti4O7和RuO2/Ti电极的析氧电位仅为2.18 V和1.47V。此外氧化剂的产生还与电极的表面性质有关。BDD电极表面吸附性差,羟基自由基主要以游离态存在[26],因此游离态的羟基自由基可以更多参与到PS的转化中,且游离出的羟基自由基空出了电极表面活性位点,从而增加了硫酸盐直接转化为PS的可能。而Ti4O7电极的表面吸附性较强,接触角小[27],羟基自由基主要以吸附态存在,容易彼此结合形成氧气,而降低了将硫酸盐转化为过硫酸盐的概率。RuO2/Ti电极虽然能在NaCl溶液中产生活性氯降解有机污染物[10],但其产生PS的能力较弱,主要与其较低的析氧电位有关。
-
图4是不同电极处理的电流和电渗流变化。由图4(a)可见,T1处理(BDD)的电流最小,T3处理(RuO2/Ti)居中,而T2处理(Ti4O7)电流最高。一般而言,电动体系的电导率越高,则电流越高。由于T1处理的BDD电极析氧电位高,水解产生的H+能力较弱,因此,产生的离子较少,电导率较低;而Ti4O7电极和RuO2/Ti电极的析氧电位均低于BDD电极,因此,产生的离子较多,体系的电导率较高,电流也较高。
电渗流的大小与电流关系密切,一般而言,电流越大则电渗流越大[28]。由图4(b)可知,电渗流均是从阳极向阴极移动(正值表示电渗流从阳极向阴极),其中T2处理的电流最高,因此其电渗流也最大。T1和T3的电流均小于T1,其电渗流也较小,分别为101.6和16.9 mL。不同电极处理的电渗流流向均从阳极向阴极,表明阳极产生的氧化剂可通过电渗流向阴极输送。
表2是不同电极处理溶液中PS浓度的变化。T1处理PS浓度为0.079~4.52 mmol·L−1,6 h时土柱各部分PS的浓度均明显高于2 h,且PS的浓度由阳极到阴极逐渐降低,表明随着处理时间的延长,BDD阳极产生的PS在电渗流的作用下逐步向阴极移动。阴极液的PS浓度也由2 h时的0.079 mmol·L−1升高到0.352 mmol·L−1,表明在电渗流作用下阳极产生的PS可以迁移至阴极池。T2中土柱各部分的PS浓度为0.079~0.195 mmol·L−1,T3中为0.079~0.211 mmol·L−1,两者差异不大,且均远低于T1处理中的PS浓度。尽管在反应池实验中,Ti4O7电极和RuO2/Ti电极在120 min时间内产生的PS浓度最高为3.27 mmol·L−1和0.379 mmol·L−1(图3(e)),但由于土柱实验中的溶液体积(1 780 mL)远高于反应池实验(240 mL),因此,产生的PS由于稀释和电渗流输送的作用导致其浓度被稀释,从而导致土柱各部分浓度较低,且未能表现出从阳极到阴极逐步递减的趋势。
PS在阳极的生成、向土柱中迁移以及作用于有机污染物的过程往往伴随着自由基的生成和转化。为探究这一系列过程,我们通过原位采集阴阳极溶液和土壤溶液,利用电子顺磁共振波谱测定生成的自由基。图5为6 h时不同阳极处理下电极液和土壤溶液自由基的EPR图谱。在6 h时,阳极液到S2中的自由基信号强度有所提高,特别是在以BDD为阳极的T1处理中,阳极液的 DMPO-OH强度最高(阳极表面可以生成·OH,式(2)),同时阳极液出现一定强度的 DMPO-SO4 信号峰(阳极表面产生的SO4·−,式(1)和(3)),表明在BDD电极附近有·OH和SO4·−的产生(图5(b))。DMPO-OH强度在T1处理的土柱中呈现阳极向阴极递减的趋势,与PS的迁移趋势类似。由于·OH的存在时间极短,不能实现由阳极向土柱的迁移行为,因此土柱中的·OH应是PS分解过程中所产生的[29-31] (式(6)~(7))。本研究中采用石英砂土柱,体系相对简单,PS产生自由基的过程为PS在迁移中先发生水解生成SO4·–,随后SO4·– 会进一步与OH−反应生成·OH,SO4·–和·OH均可降解土柱中的有机污染物蒽。在S1到阴极区并未发现明显的DMPO-SO4信号峰,原因:一是自由基捕获剂DMPO与·OH的反应速率高于SO4·−,SO4·−不容易被捕获[32],二是如前所述,SO4·−在反应过程中会持续不断地生成·OH,所以测得的SO4·−信号强度要远低于·OH。
-
表3列出了不同处理土柱中各部分溶液中PS的浓度。由于BDD阳极具有较高的自产PS氧化剂的能力,因此,T4处理中土柱各部分的PS浓度均较高,浓度为0.348~46.3 mmol·L−1。而T5和T6处理中各部分的PS浓度均较低,且浓度低于无污染物土柱实验中的浓度(表2),这主要是因为阳极产生的PS与土柱中的污染物蒽发生反应而损耗。在T4处理中,土柱各部分中的PS浓度基本遵循阳极液>S1>S2>S3>阴极液,表明在电场作用下在阳极产生的PS主要以电渗流的方式向阴极迁移[2-3,7]。由于阳极和阴极的水解作用,阳极液呈酸性,阴极液呈碱性,土柱孔隙水的pH呈现从阳极向阴极逐渐升高的规律。
图6为处理24 h后土柱各截面石英砂中的蒽的去除率。T7(对照处理)未施加直流电场,蒽的去除率为13.7%~16.5%,这是在未进行处理状态下土柱中的蒽在实验环境下挥发、光降解等影响下的自然去除率。T4处理中的蒽去除率明显高于T7,达到了64.8%~82.5%,且去除率呈现从阳极到阴极逐渐降低的趋势,这与PS在土柱中的分布规律是一致的,表明阳极产生的 PS在向阴极迁移的过程中对蒽进行了降解。这与XU等[8]、HUANG等[33]的研究结果相似,表明在EK-ISCO中氧化剂的有效输送是影响污染物去除的重要因素。T5和T6处理中蒽的去除率分别为17.4%~18.6%和17.9%~22.1%,均略高于T7,表明T5和T6产生的微量氧化剂能降解部分蒽,但由于阳极产生的氧化剂量很少,差异不显著,这与表2中氧化剂的浓度一致。
-
1)在BDD、Ti4O7和RuO2/Ti阳极材料中,BDD产PS能力最强,在使用阳离子膜、电流密度为30 mA·cm−2,Na2SO4为 0.6 mol·L−1的最优条件下,PS浓度可达到105.1 mmol·L−1。
2) BDD 阳极的处理中土柱各部分的 PS 浓度较高,且高于其他阳极处理,同时PS浓度呈现从阳极向阴极逐步降低的规律,表明阳极产生的 PS 氧化剂主要通过电渗流的方式向阴极迁移。
3) PS 在迁移的过程中,分解生成强氧化性的自由基,进而可以降解去除土柱中的污染物。在BDD 作为阳极的处理中,蒽去除率达到了64.8%~82.5%,明显高于其他2类电极的处理组。土柱各截面中蒽的去除率呈现从阳极到阴极逐步降低的规律,这与氧化剂PS在土柱中的分布规律是一致的,表明PS向土柱中的迁移是土柱中蒽去除的主要原因。
不同阳极自产氧化剂的电动迁移及对蒽的降解
Electrokinetic migration of self-produced oxidants from different anodes and their degradation of anthracene
-
摘要: 针对电动-原位化学氧化技术中需要外源投加氧化剂的问题,以3种典型阳极材料(BDD,Ti4O7和RuO2/Ti)为研究对象,以硫酸钠溶液为电解质,采用反应池和电动土柱实验,研究了不同阳极自产氧化剂的能力、氧化剂在土柱中的迁移及对有机污染物蒽的去除规律。结果表明,阳极自产氧化剂的最优条件为BDD为阳极、使用阳离子膜、30 mA·cm−2和0.6 mol·L−1的Na2SO4浓度,产生的过硫酸盐(PS)浓度最高达到105.1 mmol·L−1。在电动土柱实验中,阳极产生的PS在电渗流的作用下由阳极向阴极迁移,其中BDD阳极处理的电解液和土壤溶液中PS的浓度最高,浓度为0.079~4.52 mmol·L−1,而Ti4O7和RuO2/Ti阳极处理中PS的最高浓度仅为0.195和0.211 mmol·L−1;PS 在迁移的过程中被分解成强氧化性的自由基,从而可去除土柱中的有机污染物蒽,在BDD阳极处理中蒽的去除率可达到64.8%~82.5%,呈现从阳极到阴极逐步下降的规律,与土柱中的PS浓度分布规律一致。以上研究结果可为电动-原位化学氧化技术的发展提供技术支持。Abstract: The application of electrokinetic in situ chemical oxidation (EK-ISCO) technology has been extensively investigated for the remediation of contaminated soil. To address challenges related to high oxidant loss, exogenous oxidant addition, low migration efficiency, and limited removal efficiency of organic pollutants, it is a promising solution to use anode to generate oxidants and degrade pollutants under a direct-current electric field. In this study, the reaction cell or electrokinetic column tests were conducted for three anodes (BDD, Ti4O7 and RuO2/Ti) with a sodium sulfate electrolyte to evaluate their ability for self-produced oxidants, the migration of oxidants and the removal rules of organic pollutant anthracene in a quartz sand column. The results demonstrated that the persulfates (PS) concentration reached its peak value (105.1 mmol·L−1) under optimal conditions for anode self-produced oxidants: BDD anode, cation exchange membrane, 30 mA·cm−2, 0.6 mol·L−1 Na2SO4. In the electrokinetic column experiments, PS produced from anode could migrate to cathode under the action of electroosmosis, the PS concentrations in BDD anode treated electrolyte and soild solution reached the highest value of 0.079~4.52 mmol·L−1 , while the highest PS concentrations in the Ti4O7 and RuO2/Ti treated electrolytes were only 0.195 mmol·L−1 and 0.211 mmol·L−1, respectively. Along PS migration, they were decomposed into free radicals with strong oxidation potential to efficiently remove anthracene from the soil column. The removal rates of anthracene after BDD anode treatment ranged from 64.8% to 82.5%, and presented a gradual decline from the anode to the cathode, which was consistent with the distribution of PS concentration in the soil column. The research provides a support for the advancement of EK-ISCO technology.
-
Key words:
- anode /
- oxidants /
- persulfate /
- electrokinetic /
- anthracene
-

-
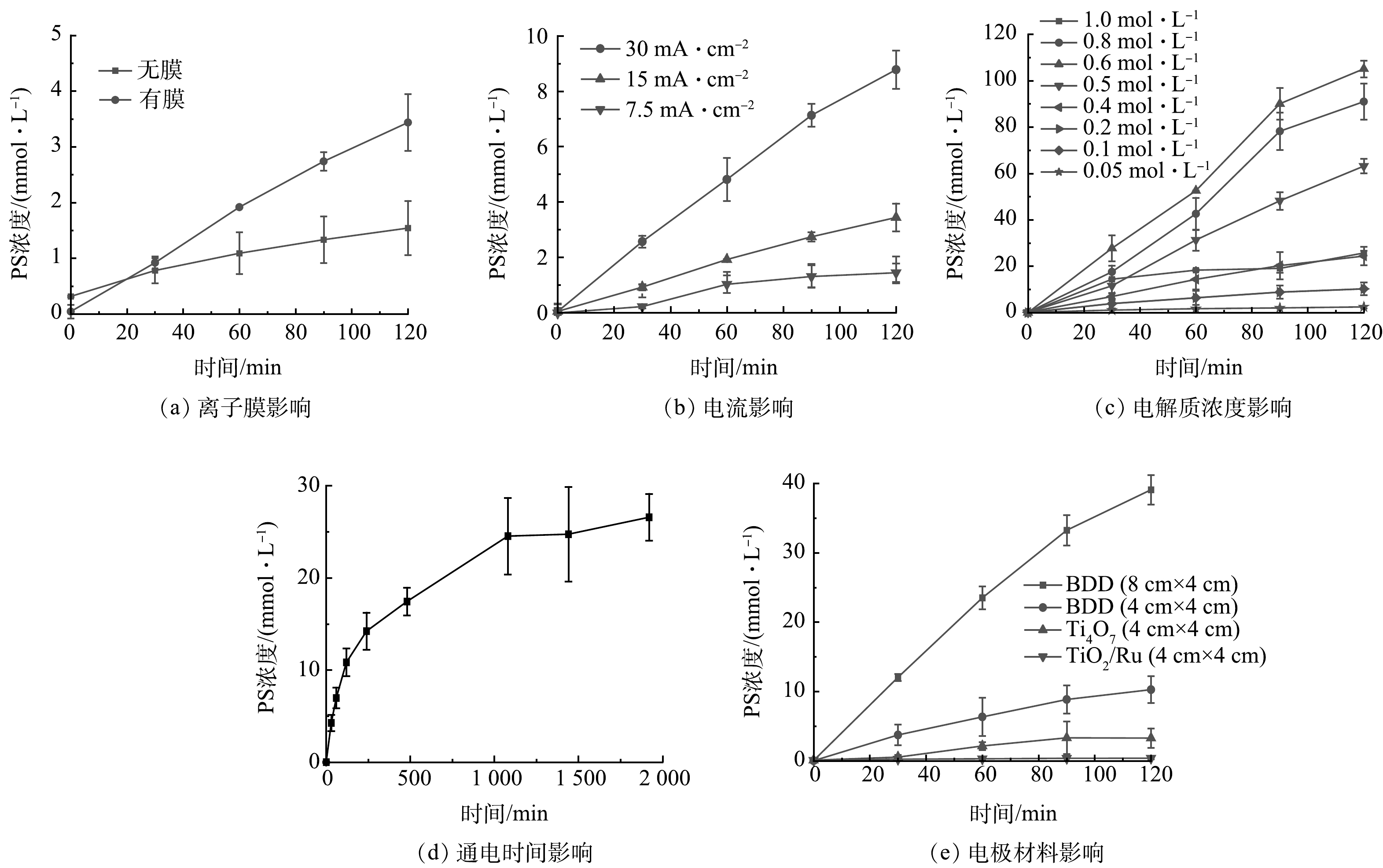
图 3 不同条件对阳极材料自产氧化剂的影响
Figure 3. Effect of different conditions on the self-produced oxidants of anode materials

图 4 不同电极处理的电流和累积电渗流随时间的变化
Figure 4. The change in current and accumulated electroosmotic flow under different electrode treatments with time
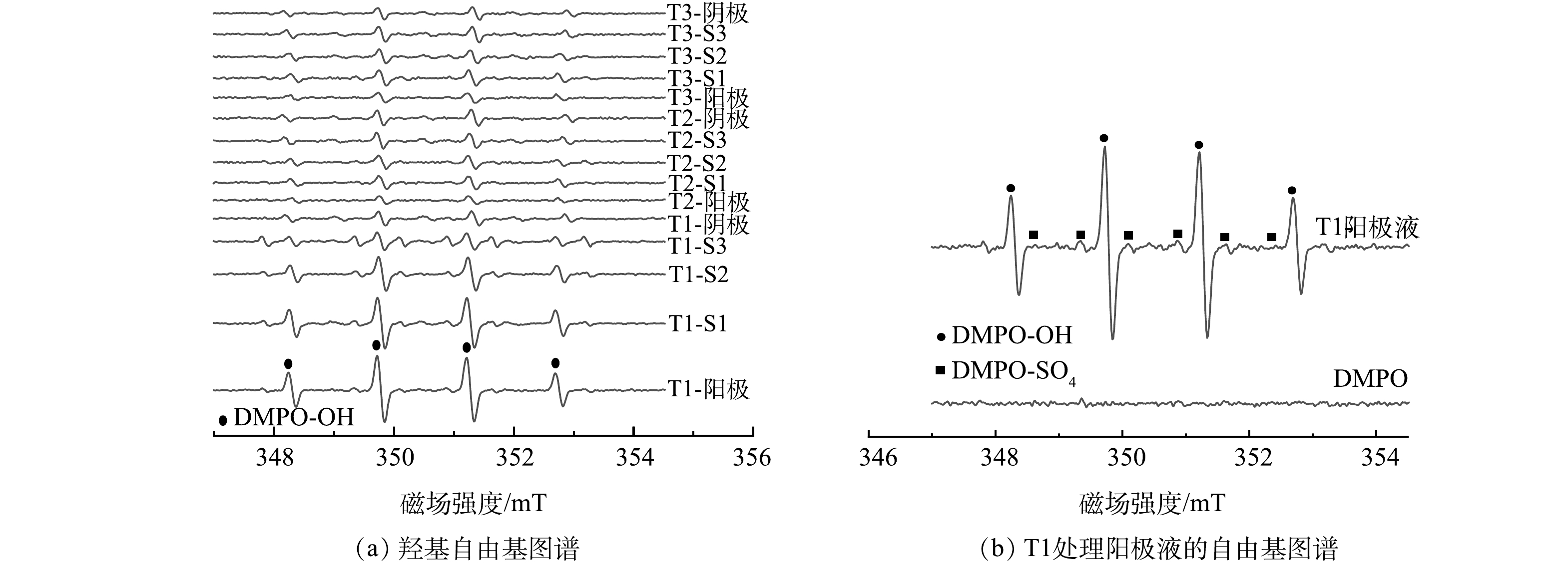
图 5 不同处理中电解液和土壤溶液的自由基图谱
Figure 5. The free radicals profiles of electrolyte and soil solution in different treatments

图 6 不同处理下土柱各截面中蒽的去除率
Figure 6. The removal rate of anthracene in different sections of quartz sand column under different treatments
表 1 不同电极自产氧化剂的影响因素实验设计
Table 1. Experimental design for influencing factors of self-generated oxidants from different electrodes
编号 阳离子交换膜 电流密度/
(mA·cm−2)Na2SO4浓度/
(mol·L−1)通电时间/min 阳极材料 1 无 15 0.1 120 BDD(4 cm×4 cm) 2 有 7.5、1、30 0.1 120 BDD(4 cm×4 cm) 3 有 30 0.05、0.1、0.2、0.4,
0.5、0.6、0.8、1.0120 BDD(4 cm×4 cm) 4 有 30 0.1 1920 BDD(4 cm×4 cm) 5 有 30 0.1 120 BDD(4 cm×4 cm)、BDD(8 cm×4 cm)、
Ti4O7(4 cm×4 cm)、RuO2/Ti(4 cm×4 cm) 下载: 导出CSV
下载: 导出CSV
表 2 不同阳极处理条件下土柱各部分溶液中的PS浓度
Table 2. PS concentrations in the solutions from various quartz column part under different anode treatments
处理 时间/h PS浓度/mmol·L−1 阳极液 S1 S2 S3 阴极液 T1 2 1.23 0.973 0.675 0.203 0.079 6 4.52 3.13 2.24 1.49 0.352 T2 2 0.186 0.203 0.079 0.095 0.169 6 0.087 0.178 0.195 0.087 0.087 T3 2 0.095 0.095 0.178 0.203 0.079 6 0.211 0.079 0.087 0.169 0.195
下载: 导出CSV
表 3 不同处理溶液中的PS浓度
Table 3. PS concentrations in the solutions from various quartz column part under different treatments
处理 时间/h PS浓度/mmol·L−1 阳极液 S1 S2 S3 阴极液 T4 6 3.04 2.96 3.04 2.31 0.348 24 46.3 25.2 14.9 11.1 6.59 T5 6 0.070 0.087 0.062 0.045 0.062 24 0.070 0.062 0.070 0.070 0.053 T6 6 0.062 0.053 0.062 0.053 0.062 24 0.070 0.053 0.045 0.053 0.062
下载: 导出CSV
-
[1] PROBSTEIN R F, HICKS R E. Removal of contaminants from soils by electric fields[J]. Science, 1993, 260: 498-503. doi: 10.1126/science.260.5107.498 [2] WEN D D, LIU H, ZHANG Y, et al. Electrokinetically-delivered persulfate and pulsed direct current electric field induced transport, mixing and thermally activated in situ for remediation of PAHs contaminated soil[J]. Journal of Hazardous Materials, 2023, 444: 130414. doi: 10.1016/j.jhazmat.2022.130414 [3] FAN G P, CANG L, GOEMES H I, et al. Electrokinetic delivery of persulfate to remediate PCBs polluted soils: Effect of different activation methods[J]. Chemosphere, 2016, 144: 138-147. doi: 10.1016/j.chemosphere.2015.08.074 [4] CANG L, FAN G P, ZHOU D M, et al. Enhanced-electrokinetic remediation of copper-pyrene co-contaminated soil with different oxidants and pH control[J]. Chemosphere, 2013, 90: 2326-2331. doi: 10.1016/j.chemosphere.2012.10.062 [5] FAN G P, CANG L, FANG G D, et al. Surfactant and oxidant enhanced electrokinetic remediation of a PCBs polluted soil[J]. Separation and Purification Technology, 2014, 123: 106-111. doi: 10.1016/j.seppur.2013.12.035 [6] ROACH N, REDDY K R. Electrokinetic delivery of permanganate into low-permeability soils[J]. International Journal of Environment and Waste Management, 2006, 1(1): 4-19. doi: 10.1504/IJEWM.2006.011122 [7] CHU L G, CANG L, FANG GD, et al. A novel electrokinetic remediation with in-situ generation of H2O2 for soil PAHs removal[J]. Journal of Hazardous Materials, 2022, 428: 128273. doi: 10.1016/j.jhazmat.2022.128273 [8] XU H T, SONG Y, CANG L, et al. Ion exchange membranes enhance the electrokinetic in situ chemical oxidation of PAH-contaminated soil[J]. Journal of Hazardous Materials, 2020, 382: 121042. doi: 10.1016/j.jhazmat.2019.121042 [9] SHEN Z M, CHEN X J, JIA J P, et al. Comparison of electrokinetic soil remediation methods using one fixed anode and approaching anodes[J]. Environmental Pollution, 2007, 150(2): 193-199. doi: 10.1016/j.envpol.2007.02.004 [10] SONG Y, CANG L, FANG GD, et al. Electrokinetic delivery of anodic in situ generated active chlorine to remediate diesel-contaminated sand[J]. Chemical Engineering Journal, 2018, 337: 499-505. doi: 10.1016/j.cej.2017.12.122 [11] 胡承志, 刘会娟, 曲久辉. 电化学水处理技术研究进展[J]. 环境工程学报, 2018, 12(3): 677-696. doi: 10.12030/j.cjee.201801179 [12] TURRO E, GIANNIS A, COSSU R, et al. Electrochemical oxidation of stabilized landfill leachate on DAS electrodes[J]. Journal of Hazardous Materials, 2011, 190(1-3): 460-465. doi: 10.1016/j.jhazmat.2011.03.085 [13] WEI W, YUAN T, YE J. Recent progress in electrochemical application of Magnéli phase Ti4O7-based materials: A review[J]. Journal of Material Science, 2023, 58: 14911-14944. doi: 10.1007/s10853-023-08929-y [14] GRGUR B N, MIJIN D Z. A kinetics study of the methomyle electrochemical degradation in the chloride containing solutions[J]. Applied Catalysis B: Environmental, 2014, 147(8): 429-438. [15] FARHAT A, KELLER J, TAIT S, et al. Removal of persistent organic contaminants by electrochemically activated sulfate[J]. Environmental Science and Technology, 2015, 49(24): 14326-14333. doi: 10.1021/acs.est.5b02705 [16] ZHOU D M, CHEN HF, CANG L, et al. Ryegrass uptake of soil Cu/Zn induced by EDTA/EDDS together with a vertical direct-current electrical field[J]. Chemosphere, 67(8): 1671-1676. [17] YANG G C C, YEH C F. Enhanced nano-Fe3O4/S2O82− oxidation of trichloroethylene in a clayey soil by electrokinetics[J]. Separation and Purification Technology, 2011, 79(2): 264-271. doi: 10.1016/j.seppur.2011.03.003 [18] FANG G D, GAO J, DIONYSIOU D D, et al. Activation of persulfate by quinones: free radical reactions and implication for the degradation of PCBs[J]. Environmental Science and Technology, 2013, 47: 4605-4611. doi: 10.1021/es400262n [19] DONG C D, CHEN C F, CHEN C W. Determination of polycyclic aromatic hydrocarbons in industrial harbor sediments by GC-MS[J]. International Journal of Environmental Research and Public Health, 2012, 9(6): 2175-2188. doi: 10.3390/ijerph9062175 [20] YUKSELEN-AKSOY Y, REDDY K R. Effect of soil composition on electrokinetically enhanced persulfate oxidation of polychlorobiphenyls[J]. Electrochimica Acta, 2012, 86: 164-169. doi: 10.1016/j.electacta.2012.03.049 [21] LI J, FU Q, LIAO Q, et al. Persulfate: a self-activated cathodic electron acceptor for microbial fuel cells[J]. Journal of Power Sources, 2009, 194(1): 269-274. doi: 10.1016/j.jpowsour.2009.04.055 [22] CHEN L, LEI C, LI Z et al. Electrochemical activation of sulfate by BDD anode in basic medium for efficient removal of organic pollutants[J]. Chemosphere, 2018, 210: 516-523. doi: 10.1016/j.chemosphere.2018.07.043 [23] YOUSEFI N, POURFADAKARI S, ESMAEILI S, et al. Mineralization of high saline petrochemical wastewater using sonoelectro-activated persulfate: Degradation mechanisms and reaction kinetics[J]. Microchemical Journal, 2019, 147: 1075-1082. doi: 10.1016/j.microc.2019.04.020 [24] DARSINOU B, FRONTISTIS Z, ANTONOPOULOU M. et al. Sono-activated persulfate oxidation of bisphenol A: Kinetics, pathways and the controversial role of temperature[J]. Chemical Engineering Journal, 2015, 280: 623-633. doi: 10.1016/j.cej.2015.06.061 [25] 卓琼芳, 杨波, 邓述波, 等. 用于有机物降解的电化学阳极材料[J]. 化学进展, 2012, 24(4): 628-636. [26] ZHU X, TONG M, SHI S, et al. Essential Explanation of the strong mineralization performance of Boron-Doped Diamond electrodes[J]. Environmental Science and Technology, 2008, 42(13): 4914-4920. doi: 10.1021/es800298p [27] WANG Q, LIU X, WANG H, et al. Ti4O7 regulating both Zn(OH)42- and electrons for improving Zn-Ni batteries[J]. Chemical Engineering Journal, 2022, 443: 136342. doi: 10.1016/j.cej.2022.136342 [28] ACAR Y B, ALSHAWABKEH A N. Principles of electrokinetic remediation[J]. Environmental Science and Technology, 1993, 27(13): 2638-2647. doi: 10.1021/es00049a002 [29] 肖鹏飞, 姜思佳. 活化过硫酸盐氧化法修复有机污染土壤的研究进展[J]. 化工进展, 2018, 37(12): 4862-4873. [30] TSITONAKI A, PETRI B, CRIMI M, et al. In situ chemical oxidation of contaminated soil and groundwater using persulfate[J]. Critical Reviews in Environmental Science and Technology, 2010, 40(1): 55-91. doi: 10.1080/10643380802039303 [31] CRIMI M L, TAYLOR J. Experimental evaluation of catalyzed hydrogen peroxide and sodium persulfate for destruction of BETX contaminants[J]. Soil and Sediment Contamination, 2007, 16(1): 29-45. doi: 10.1080/15320380601077792 [32] 彭菲, 王肖磊, 方国东, 等. 过硫酸盐在不同类型土壤中分解产生自由基的过程机制研究[J]. 土壤, 2022, 54(6): 1210-1218. [33] HUANG Q, ZHOU M Z, ZHOU J J, et al. Roles of oxidant, activator and surfactant on enhanced electrokinetic remediation of PAHs historically contaminated soil[J]. Environmental Science & Pollution Research, 2022, 29: 88989-89001. -
 点击查看大图
点击查看大图

计量
- 文章访问数: 566
- HTML全文浏览数: 566
- PDF下载数: 13
- 施引文献: 0
