

 下载:
下载:

-
在我国快速现代化进程的背景下,伴生的各类环境问题引起了社会广泛关注。其中,各类污、废水中的新污染物降解已成为目前相关领域的重点关注问题。由于新污染物具有产生来源广泛、暴露途径多样、环境风险持久等特点,对人类健康和生态安全形成了巨大的威胁[1-3]。芬顿高级氧化技术具有性能稳定、操作难度低、氧化能力强等特点,成为应用最广泛的新污染物废水处理技术[4-6]。然而,在实际操作中,Fe3+易水解和Fe2+再生效率低下的关键问题,常导致芬顿高级氧化工艺出现投药成本高、含铁危废污泥产量大等复杂现实问题,严重阻碍了技术的应用[7]。
针对以上问题,大量研究以不同的思路提升芬顿反应的氧化性能。比较典型的方法是向芬顿体系引入有机配体(如乙二胺四乙酸)[7],通过有机配体与体系中Fe3+配位,避免Fe3+的完全失活;此外,也有学者开发了盐酸羟胺助催化芬顿体系[8],利用羟胺的还原性直接强化Fe3+/Fe2+循环[9]。上述研究显著提升了芬顿工艺的氧化性能,但也存在外加试剂成本控制难度大的问题。值得注意的是,碳组分由于其还原性,在钢铁冶炼行业有极为广泛使用[10]。碳材料通常具有较高的比表面积和复杂的孔结构,相关研究[11]表明,高级氧化过程中引入碳基材料,可通过吸附、限域效应等方式实现对体系性能的促进。然而,常规反应条件下,评估碳基材料对芬顿氧化体系中Fe2+再生促进的性能和机制研究较少,使用碳材料实现芬顿反应中Fe3+/Fe2+过程的显著促进,不仅对缓解芬顿氧化的关键问题具有重要的作用,也对碳材料在环境工程领域的应用具有重大的现实意义。
本研究制备了一系列富含吡啶氮缺陷点位的碳基助催化剂(nitrogen vacancies carbon promoter,NVCx)并详细分析了助催化剂的物化性质;通过批实验分析了其助催化Fe3+/H2O2体系降解BPA过程的性能与复杂水质条件对降解过程的影响;借助顺磁共振分析、淬灭实验、电化学分析等手段对过程中形成的活性物种进行了详细鉴定;最后,通过新生态Fe2+分析、光电子能谱和量子化学计算等手段对助催化剂与Fe3+/H2O2体系间的作用机制进行了详细分析,相关结果对解决芬顿高级氧化工艺存在的现实问题和碳基材料的使用途径有全新的指导意义。
-
六水合硝酸锌(Zn(NO3)2·6H2O)、九水合硝酸铁(Fe(NO3)3·9H2O)、30%过氧化氢水溶液(H2O2,30%)、2-甲基咪唑(C4H6N2)、无水乙醇(CH3CH2OH)、甲醇(CH3OH)、乙腈(C2H3N)、叔丁醇(C4H9OH)、对苯醌(C6H4O2)、L-组氨酸(C6H9N3O2)、草酸钛钾(C4H2K2Oti)、1,10-菲罗啉(C12H8N2)等化学试剂均为分析纯,购于上海麦克林生化科技股份有限公司,实验用水均出自Milli-Q 超纯水系统。
-
1)助催化剂的制备。使用ZIF-8作为助催化剂制备所需的前驱体,制备过程如下:称取4.13 g六水合硝酸锌和4.40 g二甲基咪唑分别分散于100 mL甲醇中,将分散好的二甲基咪唑溶液迅速加入六水合硝酸锌溶液中,充分搅拌反应12 h,静置反应4 h后离心。将所得白色沉淀物质用乙醇快速清洗2遍后置于70 ℃烘箱中烘干,得到白色粉末状ZIF-8。将ZIF-8粉末置于刚玉瓷舟中,在N2气氛保护下置于管式炉中分别在800、900、1 000 ℃等目标温度下热解3 h(升温速率为10 ℃·min−1),将得到的黑色粉末置于1.0 mmol·L−1硫酸溶液中热浴反应4 h后持续水洗,直至上清液pH呈中性,烘干即得碳基助催化剂NVC800、NVC900和NVC1 000 (下标代表制备过程的热解温度)。
2)实验仪器。使用Gemini SEM 500场发射扫描电镜(SEM)观察前驱体和助催化剂的微观形貌;使用Rigaku Ultima Ⅳ型X射线衍射仪(XRD)确定前驱体及助催化剂的物相;使用Thermo Fisher K-Alpha型X 射线光电子能谱仪(XPS)分析反应组分的价态环境;使用DXR 2Xis型拉曼光谱仪(Raman)分析助催化剂的缺陷度;使用Nicolet iS50型傅里叶红外光谱仪(FTIR)分析助催化剂的碳结构振动模式;使用Bruker EMXmicro-6/1电子顺磁共振波谱仪(EPR)分析助催化剂结构缺陷伴生的未成对电子与氧化反应过程中生成的活性物种类型;使用电化学工作站(CHI660E)分析H2O2活化过程中助催化剂与Fe组分之间的电荷迁移行为,测试过程使用三电极体系,以银-氯化银电极作为参比电极,以铂电极作为对电极,以碳玻电极作为工作电极,电解质采用0.1 mol·L−1硫酸钠溶液。
3) 降解实验。使用120 mL烧杯开展污染物降解实验,实验过程如下:将BPA溶液置于烧杯中,使用HNO3(0.1 mol·L−1)或NaOH(0.1 mol·L−1)将溶液pH调至低于3.0,随后加入一定量预配置的Fe3+储备液,将溶液定容至100 mL并同时调整溶液pH至4.0±0.1。将一定质量助催化剂加入混合溶液中,超声分散10 s,使用精密移液枪加入一定量H2O2以激活氧化反应。在不同的预设时间取样,样品使用0.22 μm滤膜过滤,置于高效液相色谱样品瓶中并使用0.5 mL甲醇进行淬灭。如无特殊声明,常规实验条件如下:实验温度为(25±2) ℃,BPA浓度为60 μmol·L−1,Fe3+浓度为0.04 mmol·L−1,H2O2浓度为5.0 mmol·L−1,催化剂投加质量浓度为0.1 g·L−1,溶液初始pH为4.0±0.1。所取样品在2 h内通过高效液相色谱仪(岛津LC-20A)进行检测,流动相采用纯水和乙腈的混合液(乙腈体积比为50%),检测波长为278 nm,流动相流速为1.0 mL·min−1,进样体积为10.0 μL,样品仓和柱温箱保持35 ℃恒定。其他污染物的浓度测定的条件如下:磺胺甲恶唑(sulfamethoxazole,SMX)的测定采用纯水和乙腈混合液作为流动相(乙腈体积比为40%),检测波长为270 nm;对乙酰氨基酚(acetaminophen,AAP)和阿特拉津(atrazine,ATZ)均使用纯水和甲醇的混合液作为流动相(甲醇体积比分别为20%和70%),检测波长分别为248 nm和225 nm;甲基橙(Methyl Orange,MO)和亚甲基蓝(Methylene blue,MB)使用紫外分光光度计进行测量,其检测波长分别为664 nm和465 nm。
-
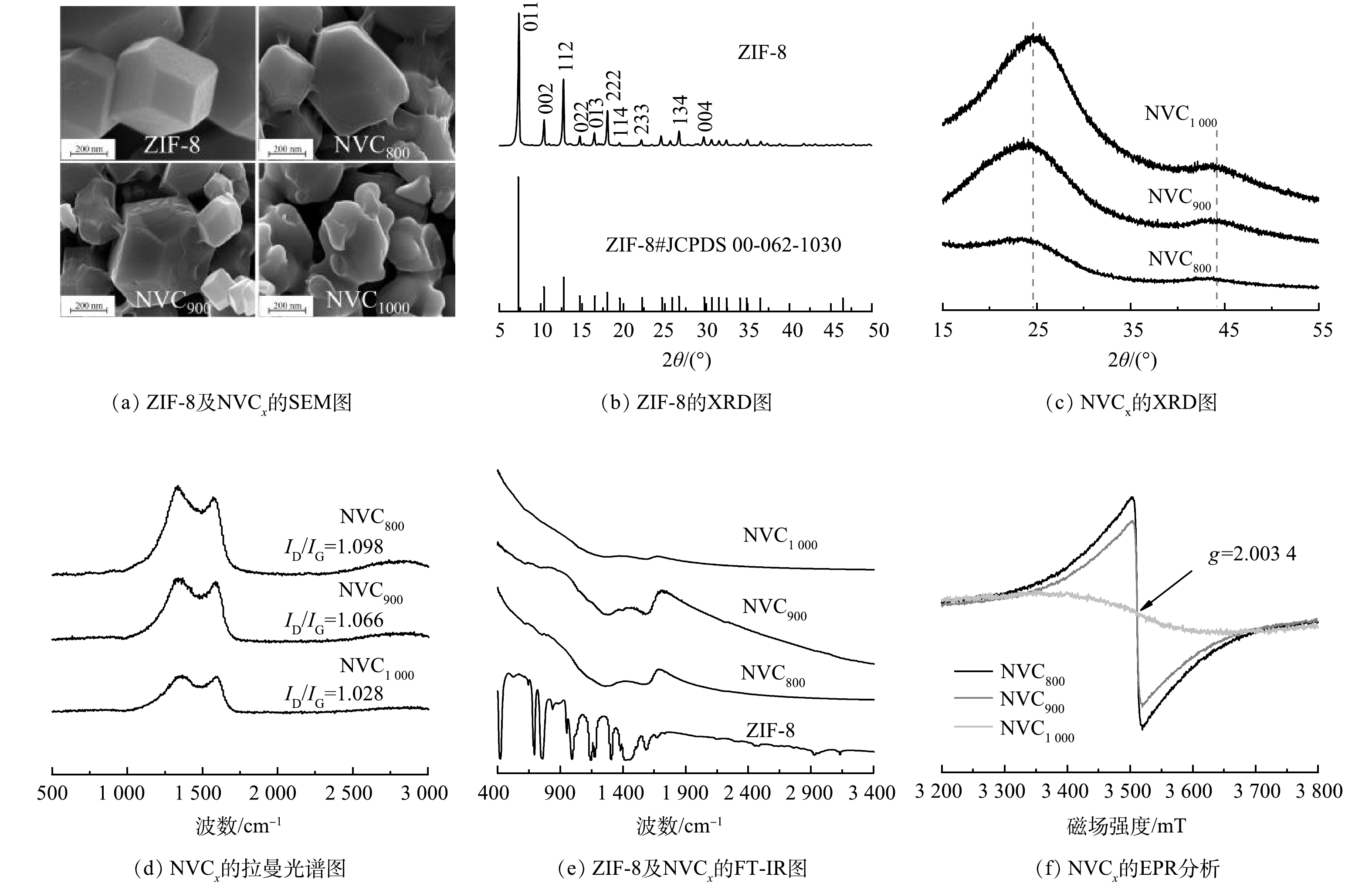
首先,对ZIF-8前驱体及不同温度下制备的助催化剂的物化性质进行了初步分析。各样品的SEM分析结果如图1(a)所示,可以发现,前驱体ZIF-8呈边缘清晰的正十二面体晶体结构,与现有研究[12]所报道的ZIF-8形貌特征一致。通过ZIF-8的限氧热解制备助催化剂,当热解温度由800 ℃升高至900 ℃时,ZIF-8的物理结构由于Zn的熔融和碳基配体的碳化发生显著的变化,但仍然保留了ZIF-8的形貌特征。当进一步将热解温度提升至1 000 ℃时,由于Zn的汽化和N2的持续吹扫,碳化产物基本失去了ZIF-8的形貌特征,其颗粒尺寸约为200~400 nm。
进一步对各样品的物相进行了分析,相关XRD分析结果见图1(b)与(c)。可以看出,前驱体ZIF-8的衍射结果较为复杂,分别在2θ为7.30、10.35、12.70、14.80、16.40和18.00°的位置出现了显著的衍射峰,且晶化度较高。上述衍射峰分别对应ZIF-8(JCPDS 00-062-
1030 )晶体的(011)、(002)、(112)、(022)、(013)和(222)晶面[13]。对于ZIF-8热解后的3种产物(NVCx),在扫描区间为15.0~55.0°内,NVC800、NVC900和NVC1 000仅在24.7°和43.7°出现2个半峰宽极大的峰,分别对应石墨碳的(002)和(100)晶面[14],表明在热解阶段前驱体发生了碳化,而较宽的半峰宽也表明所得碳化产物的晶化度较低,属于短程有序的碳材料。同时,金属相关衍射峰的缺失表明前驱体中Zn在热解和酸洗过程中被完全去除。随后,对NVCx的Raman光谱进行了分析,结果见图1(d)所示。各NVCx在1 360 cm−1和1 580 cm−1处均出现较强的特征峰,分别对应碳材料缺陷振动(D band)与碳原子sp2杂化的面内伸缩振动(G band)。两峰强度比ID/IG常用来量化碳材料的缺陷程度[15],相关计算结果表明NVCx缺陷程度随热解温度升高逐渐降低,表明高温条件可实现碳材料的结构重构。
进一步地对前驱体ZIF-8和NVCx的FT-IR光谱进行了测试分析。如图1(e)所示,前驱体ZIF-8的红外光谱较为复杂,样品热解后,随着有机配体分子的碳化,其红外光谱得到了大幅度的简化,ZIF-8样品中421 cm−1处的Zn—N振动峰以及400~1 400 cm−1内关于ZIF-8的一系列特征振动谱带在热解过程中均完全消失[16],同时,热解样品中的C—C振动强度逐渐增大[17]。对于NVCx,1 580 cm−1处的峰对应碳共轭结构面内伸缩振动,当热解温度达到1 000 ℃后,该峰的强度大幅度降低。
最后,对NVCx进行了电子顺磁共振分析,结果如图1(f)所示。可以发现,NVCx的洛伦兹线中心均位于g=
2.0034 处,对应于氮杂质缺陷结构中的未成对π电子[18],表明助催化剂具有电子供出的潜力。同时,可以发现随着热解温度的提升,洛伦兹线宽度逐渐变宽,且峰强度有所降低,表明氮缺陷不断减少,该结果与拉曼光谱和红外光谱分析结果一致,同时也说明掺杂态的氮元素在高温条件下不稳定。 -
选择双酚A(bisphenol A,BPA)作为特征污染物,验证常规实验条件下(具体见1.2)NVCx的助催化性能,结果如图2(a)所示。可以看出,在不同助催化剂共存的Fe3+/H2O2反应体系中,BPA均可在10 min内实现完全降解。其中,NVC800、NVC900和NVC1 000在助催化Fe3+/H2O2降解BPA过程中的反应动力学常数分别为0.362 0、0.373 0和0.273 9 min−1;作为对照,仅Fe3+与H2O2共存的条件下,BPA降解反应动力学常数为0.163 2 min−1,表明助催化剂对Fe3+/H2O2体系有良好的促进效果。需要指出的是,NVC900在该过程中表现出了强于NVC800和NVC1 000的助催化性能。同时,NVC900/H2O2和NVC900/Fe3+体系对BPA的降解率均小于5%,表明NVC900无法独自实现H2O2的活化,且助催化剂对BPA的吸附作用并非导致BPA高效降解的主要原因。进一步考察了芬顿反应(Fe2+/H2O2)体系与NVC900助催化芬顿反应体系(NVC900/Fe2+/H2O2)中的BPA降解行为,结果表明BPA在上述2个体系中的降解动力学常数分别达到了1.086 9 min−1和1.805 2 min−1。该结果表明NVC900的共存亦可显著助催化芬顿反应,实现H2O2的高效活化,同时也表明助催化剂的投加可能对Fe3+的水解有抑制效果或对Fe3+/Fe2+价态循环有一定的促进。因此,为独立评价其助催化特性、排除初次投加Fe2+的原生催化活性,后续研究使用芬顿反应中的失活催化组分(Fe3+)开展实验。
其次,对NVC900助催化Fe3+/H2O2体系的最佳投药量进行了单一影响因素的实验研究,结果如图2(b)~(c)所示。在常规反应条件下,BPA降解反应动力学常数随着Fe3+浓度的提高而提高。当Fe3+的浓度为0.01 mmol·L−1时,BPA的降解速率常数为0.114 7 min−1。当Fe3+浓度增加至0.04 mmol·L−1时,降解速率常数达到0.373 0 min−1。当进一步增加Fe3+投加量至0.08 mmol·L−1时,BPA降解的动力学常数并未随之提高,反而出现了轻微的降低,表明NVC900助催化Fe3+/H2O2体系在较高Fe3+浓度时受到一定的抑制。造成该结果的原因可能是较高Fe3+浓度消耗了部分H2O2以还原自身,导致了污染物降解速率的降低(式(1)),此外,过量的Fe3+难以与助催化剂接触是可能的另一原因。此外,BPA降解速率受过量H2O2投加的影响。当H2O2投加量由5.0 mmol·L−1分别增至10.0 mmol·L−1时和20.0 mmol·L−1时,BPA的降解速率并未随之上升,反而分别下降至0.230 6 min−1和0.278 4 min−1。造成该结果的原因可能来自多方面,其中过量H2O2对羟基自由基(·OH)的淬灭导致活性物种的无效消耗是目前公认的原因(式(2))[19],该结果也说明·OH可能是NVC900催化Fe3+/H2O2体系中主要的活性物种。
第三,在常规反应条件下,考察了不同初始pH时NVC900助催化Fe3+/H2O2体系的氧化性能(图2(d))。结果表明,当溶液pH分别为3.0、3.5和4.0时,NVC900助催化Fe3+/H2O2氧化体系可在20 min内实现对BPA的完全降解,并在溶液pH为4.0时达到最高的降解速率。上述结果表明,弱酸条件下NVC900助催化的Fe3+/H2O2体系对BPA有极强的氧化性能。值得注意的是,在pH为2.0和5.0条件下,20 min内BPA在NVC900/Fe3+/H2O2体系降解率分别为60%和25%,该降解效率的下降由Fe3+与助催化剂的接触、H2O2一级解离等因素共同造成,但相比于同条件下Fe3+/H2O2体系27%和18%的BPA降解率,NVC900的助催化效果在较宽pH范围内仍较为显著。
进一步地,考察了常规反应条件下共存离子对NVC900助催化Fe3+/H2O2体系性能的影响,结果如图2(e)所示。可以看出,溶液中分别共存0.15 mmol·L−1的Cl−和HCO3−时,NVC900助催化Fe3+/H2O2体系降解BPA的性能受到轻微抑制,降解动力学常数分别下降至0.331 3 min−1和0.339 5 min−1。其中,Cl−的共存易消耗·OH,产生氧化性较弱的Cl·和Cl2·−(式(3~5))[20],造成体系氧化性能下降,而HCO3−易消耗体系酸度,并竞争·OH生成碳酸根自由基是其抑制体系氧化性能的原因(式(6));此外当溶液中共存0.15 mmol·L−1的H2PO4−时,该体系的催化性能被明显抑制,而H2PO4−易使Fe3+沉淀导致催化组分的彻底失活,是其抑制体系氧化性能的直接原因(式(7))。而当基准反应条件下存在0.15 mmol·L−1 NO3−时,BPA的降解速率未发生明显变化。
为进一步考察NVC900的助催化性能,分析了NVC900助催化的Fe3+/H2O2体系对其他污染物的降解性能,在常规反应条件下,分别选择SMX、ATZ、AAP、MB和MO作为目标污染物进行降解。结果表明,在20 min内SMX、AAP、MB和MO均可实现完全降解,ATZ虽未被完全降解,但其降解效率仍接近90%,表明NVC900助催化的Fe3+/H2O2体系对多种类型的有机污染物均可保持良好的污染物降解效率。
最后,为了考察NVC900的助催化活性的稳定性,在常规反应条件下进行了低浓度的(6.0 μmol·L−1)BPA降解循环实验,结果如图3所示。助催化反应后的NVC900经甲醇清洗-硫酸清洗-纯水清洗-烘干的方法再生后进行了连续5轮的助催化实验,待测体系TOC去除率由首轮的68.76%逐步下降至第五轮的57.54%,表明NVC900拥有较强的循环使用性能。更重要的是,作为新污染物的BPA在水体中常以极低浓度出现,低浓度下较低的反应驱动力使其很难去除,而本研究中的NVC900催化Fe3+/H2O2体系不仅可将BPA氧化为小分子有机物,而且很大程度上实现了对其中间产物的矿化。
上述结果表明,NVC900助催化Fe3+/H2O2体系在较宽pH范围和较多种离子共存时对BPA可实现很强的氧化及矿化能力,并在使用过程中展现出较强的循环稳定性。此外,该体系还可对多种新污染物表现出优良的降解能力,其过程机制值得深入研究。
-
高级氧化过程中,难降解有机污染物的去除主要通过各类活性物种的行程而实现。为了解NVC900助催化Fe3+/H2O2体系中的活性物种类型及产生机制,首先以5,5-二甲基-1-吡咯啉-N-氧化物(5,5-dimethyl-1-pyrroline N-oxide,DMPO)与2,2,6,6-四甲基哌啶(2,2,6,6-tetramethylpiperidine,TEMP)作为自旋捕获剂[21],通过EPR分析对体系反应过程中的活性物种进行了识别,结果见图4(a-c)。
可以发现,在水溶液体系中,DMPO可捕获到峰强比例1:2:2:1的信号,为羟基加成物DMPO-·OH的特征峰。而在甲醇作为溶剂体系中,DMPO可捕获到一组峰强相等的六线峰,为典型的超氧加成物DMPO-O2·−特征峰。此外,在水溶液体系中,TEMP也可捕获到强度较弱的三线峰,是典型的单重态氧存在的信号。值得注意的是,DMPO-·OH的信号强度随反应时间先增强后减弱,表明·OH在NVC900助催化Fe3+/H2O2体系中大量生成。DMPO-O2·−在反应开始后5 min内达到最大值,并在后续反应中保持稳定强度,表明O2·−在反应过程中同样有显著的生成,其可能的产生路径如(式(8))[22]。TEMP-1O2信号始终呈现较弱强度,可能是由于O2·−的歧化反应生成了极少量1O2(式(9))[23]。
进一步通过淬灭实验验证EPR分析结果的准确性,通常来说,目前相关研究倾向于选用甲醇作为·OH淬灭剂、对苯醌作为O2·−淬灭剂以及L-组氨酸作为1O2淬灭剂。由图4(d)结果可知,各淬灭剂均对体系产生了明显的淬灭效应。但值得注意的是,对苯醌和L-组氨酸不仅可有效淬灭O2·−与1O2,其各自与·OH的反应速率常数分别为1.2×109 L·(mol·s)−1与5.0×109 L·(mol·s)−1,甚至高于甲醇与·OH的反应速率常数(9.7×108 L·(mol·s)−1)[24]。通过对比不同淬灭剂与各活性物种的反应速率,可以推定·OH为体系主要活性物种。
最后,铁组分参与的高级氧化过程中常出现的高价铁活性物种([FeIV=O]2+)[25](式(10))。由于甲基苯基亚砜(phenyl methyl sulfoxide, PMSO)可被[FeIV=O]2+选择性氧化为甲基苯基砜(phenyl methyl sulfone, PMSO2),因此,选择PMSO作为判断[FeIV=O]2+是否生成的化学探针。本研究使用该方法对体系中可能的高价铁进行了分析,结果见图5(a)。在初始PMSO浓度为50 μmol·L−1时,10 min内Fe3+/ H2O2体系对PMSO降解率达18%,体系中未发现PMSO2的生成,表明Fe3+/ H2O2体系中未生成[FeIV=O]2+活性物种。在相同条件下加入NVC900后,10 min内PMSO降解率增至30%,表明NVC900助催化下Fe3+/ H2O2体系产生了更多氧化活性物种,该现象与顺磁共振结果(图4(a-c))一致。同时,PMSO2在相同时间内生成率不足1%,表明[FeIV=O]2+并未在NVC900助催化的作用下生成。
最后,通过电化学测试考察了NVC900催化Fe3+/H2O2体系中的电子转移途径,结果见图5(b)。向负载NVC900的工作电极所构建的电解体系施加0.29 V的恒电位时(由电化学开路电压确定),测得电解体系恒电位电流为−3.8×10−7 A。待电流稳定后首先向体系加入0.1 mmol·L−1的Fe3+,电流升高降至−3.79×10−6 A,表明Fe3+在NVC900表面发生了电化学吸附并得到电子,与顺磁共振分析所得推论一致。进一步向体系中加入2.0 mmol·L−1的H2O2,电流进一步升高至−4.8×10−6 A,表明H2O2的共存消耗了电子。
-
为进一步探究NVCx助催化Fe3+/H2O2过程的机制,利用1-10菲罗啉法对不同条件下Fe组分的价态变化进行了详细分析[26]。首先,在pH为4.0,Fe3+浓度为0.04 mmol·L−1,NVC900投加质量浓度为0.1 g·L−1、H2O2浓度为5.0 mmol·L−1条件下分析了Fe2+再生过程的特点,结果见图6(a)。可知,60 min的反应时间内, Fe3+/ NVC900、Fe3+/H2O2和NVC900/Fe3+/H2O2体系中的Fe2+累积转化率分别达到了31%、34%和73%。该结果表明,在NVC900助催化芬顿反应的过程中,新生态Fe2+的生成主要分为2个主要途径:其一,吸附于助催化剂表面的Fe3+在助催化剂作用下被还原为Fe2+;其二,共存的H2O2亦可还原Fe3+从而产生新生态Fe2+。
随后,利用草酸钛钾分光光度法考察了NVC900 /Fe3+/ H2O2体系在60 μmol·L−1 BPA 条件下H2O2利用率[27],结果如图6(a)所示。在8 h的反应时间内,H2O2利用率为46%。以上结果表明,Fe3+被NVC900吸附还原为Fe2+后引发芬顿氧化中的链式反应,在提高过氧化氢利用率的同时伴随着高效的活性物种释放,从而对污染物降解产生显著的促进作用。NVC800、NVC900和NVC1 000的XPS氮元素(N1s)精细谱解卷积分析结果如图6(c)所示,吡啶氮占比分别为61.68%、62.54%和61.39%,是导致Fe3+/Fe2+循环性能加强的潜在原因。
随后,使用XPS进一步验证了Fe3+吸附于NVC900表面后铁元素的价态信息,结果如图6(c)所示。Fe(NO3)3作为Fe3+的标准物,仅于725.6 eV和712.5 eV结合能处出现特征双峰,分别对应Fe3+的2p1/2轨道和2p3/2轨道。助催化Fe3+/H2O2体系后的NVC900同样出现了Fe3+的特征双峰,更重要的是,710.5 eV和723 eV出现了Fe2+的特征双峰,对应于NVC900表面还原后未释放入溶液体相中的Fe2+,该结果与图6(a)中Fe2+再生实验结果一致,再次验证了NVC900加速Fe3+/ Fe2+循环的相关结论。最后,考察了NVC900-Fe在活化H2O2前后表面氮元素的组分变化,结果见图6(d)。由图可见,反应前后NVC900-Fe中吡啶氮的含量几乎未发生衰减,表明吡啶氮电子并非Fe3+还原的直接电子来源。以上结果表明,当NVC900加入Fe3+溶液后,NVC900热解过程中产生的大量吡啶氮缺陷可迅速吸附溶液中的Fe3+,并在一定反应时间后于NVC900表面有效吸附Fe3+并实现新生态Fe2+的再生[28],再生机制值得进一步研究。
-
最后,本研究使用B3LYP方法,在6-31+G(d,p)的全电子基组下进行密度泛函计算,对NVC900助催化Fe3+活化H2O2过程的详细机制进行了计算分析。计算过程使用Gaussian 16 W C.01量子化学软件[29],借助Gaussview 6.0与Multiwfn软件进行可视化数据分析[30]。
根据XPS分析结果,对助催化剂吸附Fe3+过程进行计算,结果见图7(a)。可以发现,NVC900对Fe3+的吸附能和NVC900-Fe对H2O2的吸附能分别为−3.06 eV和−2.14 eV,表明NVC900-Fe的形成与进一步的H2O2吸附为自发过程。进一步探究了H2O2在吸附前后O—O键长度变化。结果表明H2O2吸附于NVC900-Fe铁原子处后,O—O键延长了5.3%,表明H2O2被吸附后,其分子结构发生变化,为后续活化产生羟基自由基提供了先决条件。此外,Fe—N键键长为0.199 2 nm,与相关研究的结果[31]极为接近。最后,分析了上述过程中的电荷转移方向,发现Fe3+吸附于助催化剂的氮缺陷处以后,铁中心的电荷布居(NBO电荷)由+3降低至+1.1,该结果与Fe2+再生实验、电化学实验及XPS表征结果完全一致,直接证明了助催化剂对Fe3+/Fe2+循环过程的促进机制。在吸附H2O2后,铁中心电荷布局电荷升高至1.25 4,同时H2O2的电荷由0降低至−0.197 0,表明NVC900-Fe活化H2O2过程中向H2O2供出了电子,从而实现其活化[28]。同时,对NVC900吸附Fe3+过程前后的π电子布居进行了详细分析,结果如图7(b)所示。可以发现,氮掺杂的碳助催化剂中存在丰富的π电子,当Fe3+吸附于氮缺陷处后,助催化剂中的π电子丰度显著降低,而在中心铁离子处出现了显著的电荷,表明Fe3+吸附于助催化剂的氮缺陷处发生Fe2+再生现象的主要原因是助催化剂中的π电子传递,进而实现共存H2O2的高效活化。基于此,可以合理推测的是,尽管助催化剂在5轮的连续评估中展现出了良好的助催化剂活性,但该助催化过程难以在较长的时间内持续生效,其助催化活性容量与π电子消耗直接相关,该推论与连续使用过程中TOC去除效率缓慢降低的趋势一致。
-
本研究以ZIF-8为模板,通过限氧热解制备了一系列碳基助催化剂,以BPA作为特征污染物探究了其助催化Fe3+/H2O2的最佳反应条件及过程机制,主要结论如下:
1)在反应温度为(25±0.5) ℃、Fe3+浓度为0.04 mmol·L−1、初始pH为4.0、助催化剂投加质量浓度为0.1 g·L−1、H2O2浓度为5.0 mmol·L−1条件下,900 ℃条件下制备的助催化剂(NVC900)具有最强的助催化活性,其助催化Fe3+/H2O2体系对BPA降解过程的反应速率达到了0.373 0 min−1。
2) NVC900可对溶液中的Fe3+产生吸附作用,并实现表面吸附态Fe3+的还原,从而导致新生态Fe2+的持续生成。该过程保证了体系中共存H2O2的持续活化和活性物种的释放,体系在连续5轮的使用过程中均对共存BPA保持了良好的矿化效率。
3)活性物种识别分析表明,羟基自由基(·OH)、超氧自由基(O2·−)与单重态氧(1O2)均可在NVC900助催化Fe3+/H2O2过程中识别到,其中,羟基自由基对污染物降解起主要作用。H2O2与NVC900-Fe接触过程中的O—O键延长与接受电子是H2O2活化的主要原因。H2O2的活化主要通过O—O的均裂,体系中并未检测到高价铁物种的生成。
氮杂质缺陷碳助催化剂增效类芬顿反应过程机制
Mechanism of nitrogen impurity defect carbon promoter enhanced fenton-like reaction process
-
摘要: 本研究以ZIF-8为模板制备了一系列氮掺杂碳材料,分析其对Fe3+/H2O2体系的助催化性能与作用机制。使用X射线衍射仪、傅里叶红外光谱仪、拉曼光谱仪、X射线光电子能谱仪等手段,对助催化剂物相、形貌特征以及化学组分进行了详细分析。结果表明,所制备的助催化剂具有丰富的吡啶氮类杂质缺陷点位,且缺陷丰度随助催化剂制备温度的升高而下降;污染物降解实验表明,助催化剂的投加可显著增强Fe3+向Fe2+的还原,从而强化芬顿氧化体系对共存有机物的降解性能;顺磁共振分析、活性物种淬灭与电化学等实验表明,反应体系中共存有·OH、·O2−、1O2等活性物种,其中,·OH对共存污染物的降解起主导作用;通过X射线光电子能谱分析与量子化学计算对助催化过程机制进行了详细分析,结果表明,芬顿反应过程中失活催化组分(Fe3+)在助催化剂氮缺陷处的吸附伴随了由助催化剂向吸附态Fe3+的电子传递过程,是Fe2+再生的主要原因。以上研究结果可为缓解芬顿反应中Fe3+/Fe2+循环效率低的关键问题提供全新的思路,对碳基环境功能材料与芬顿氧化技术的耦合与应用可提供数据支撑。Abstract: This study prepared a series of nitrogen-doped carbon materials by using ZIF-8 as a template to investigate their promotion performance and catalytic mechanism toward Fe3+/H2O2 system. The physical phase, morphological characteristics, and chemical composition of the promotor were analyzed with X-ray diffraction, Fourier transform infrared spectroscopy, Raman spectroscopy, and X-ray photoelectron spectroscopy. The results showed that the prepared promotor was abundant in pyridinic-nitrogen extrinsic defects, and the defect degree decreased with the increase of pyrolysis temperature during synthesis. Pollutant degradation experiments demonstrated that the addition of the promoter significantly facilitated the regeneration of Fe2+ from Fe3+, and strengthened the performance of Fenton matrix on the degradation of coexisting organic compounds. Electron paramagnetic resonance analysis, reactive oxidation species (ROS) quenching, and electrochemical experiments indicated that ROS such as ·OH, ·O2−, and 1O2 were involved in the reaction system, of which ·OH played a major role. Detailed analysis of the promotion mechanism was conducted through X-ray photoelectron spectroscopy and quantum chemical calculations. The results indicated that the adsorption of Fe3+ onto nitrogen defects on the surface of the promoter was accompanied by an electron transfer process from the promoter to adsorbed Fe3+, which was responsible for the regeneration of Fe2+. These findings provided a novel approach to addressing the key issue of low Fe3+/Fe2+ cycling efficiency in the Fenton reaction, and provide a theoretical and data support for the coupling and application of carbon-based environmental functional materials with Fenton oxidation technology.
-
Key words:
- promoter /
- Fenton oxidization /
- carbon defect /
- reactive oxidation species
-

-
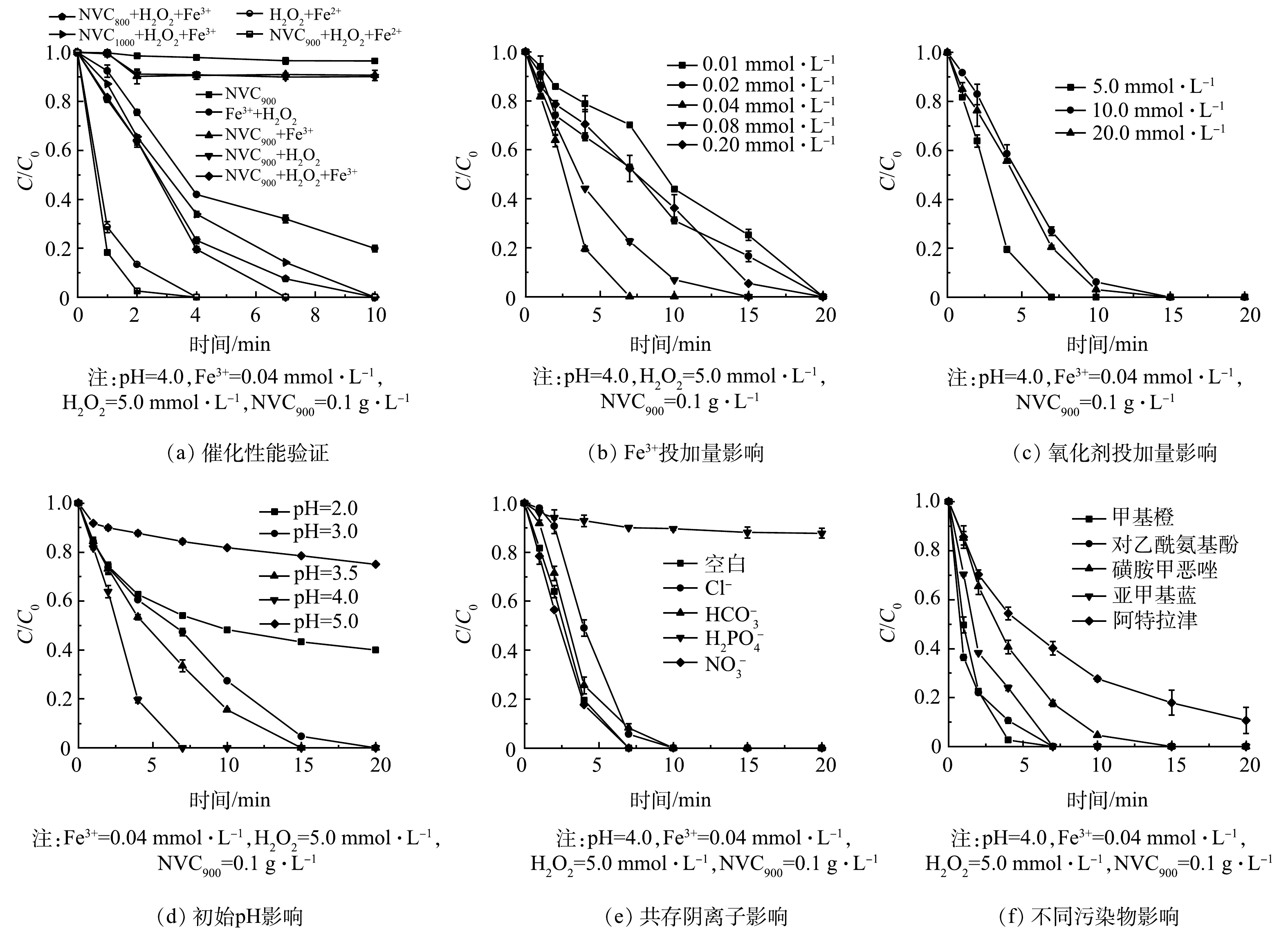
图 2 NVCx的助催化Fe3+/H2O2体系降解BPA过程的性能与影响因素分析
Figure 2. Performance and influencing factors analysis of NVCx-promoted catalytic degradation of BPA in Fe3+/H2O2 system
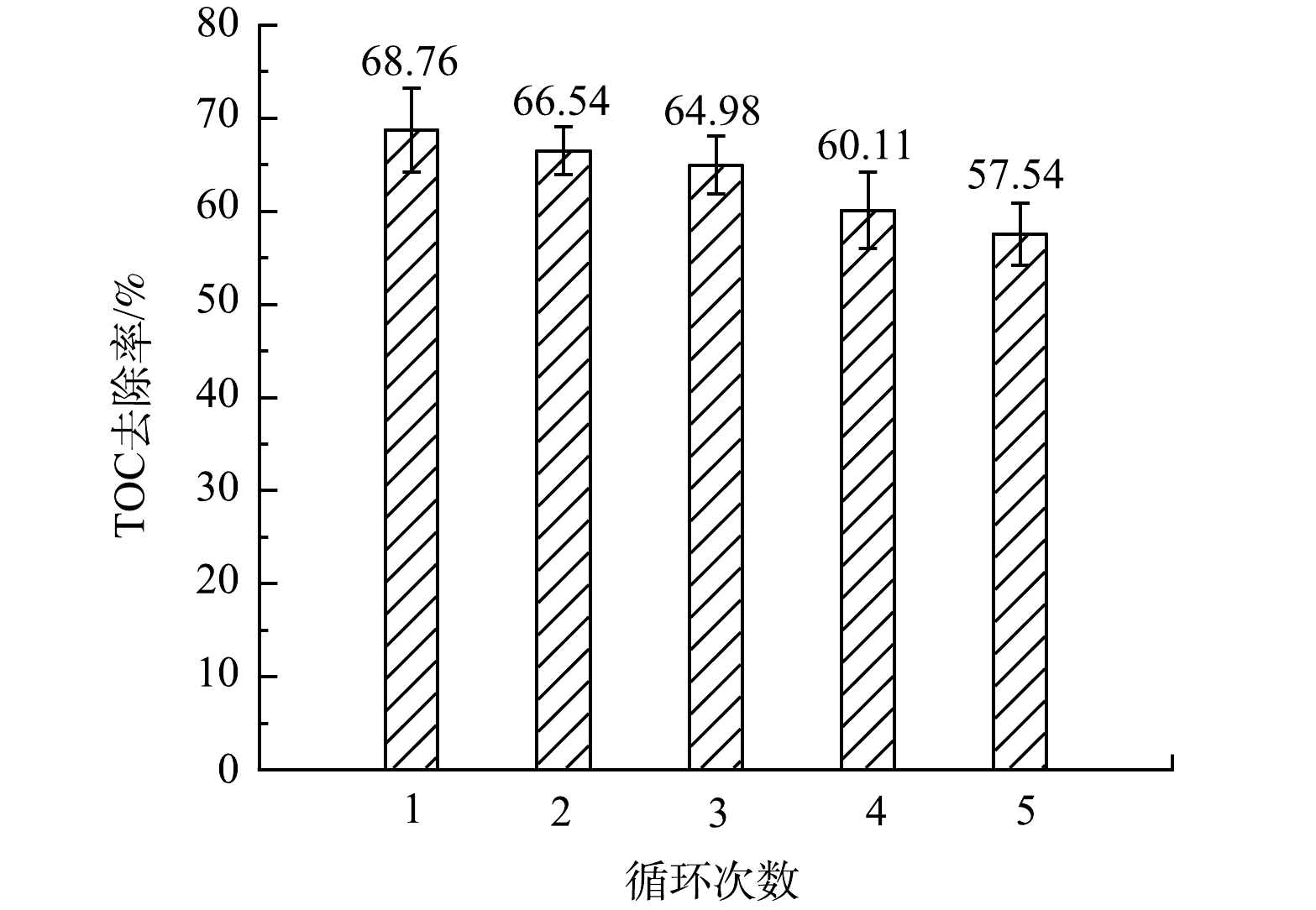
图 3 NVC900助催化Fe3+/H2O2体系的循环实验分析
Figure 3. Cyclic experimental analysis of NVC900 catalytic Fe3+/H2O2 system

图 4 NVC900/Fe3+/H2O2体系顺磁共振及淬灭实验
Figure 4. EPR and quenching experiment of NVC900/Fe3+/H2O2
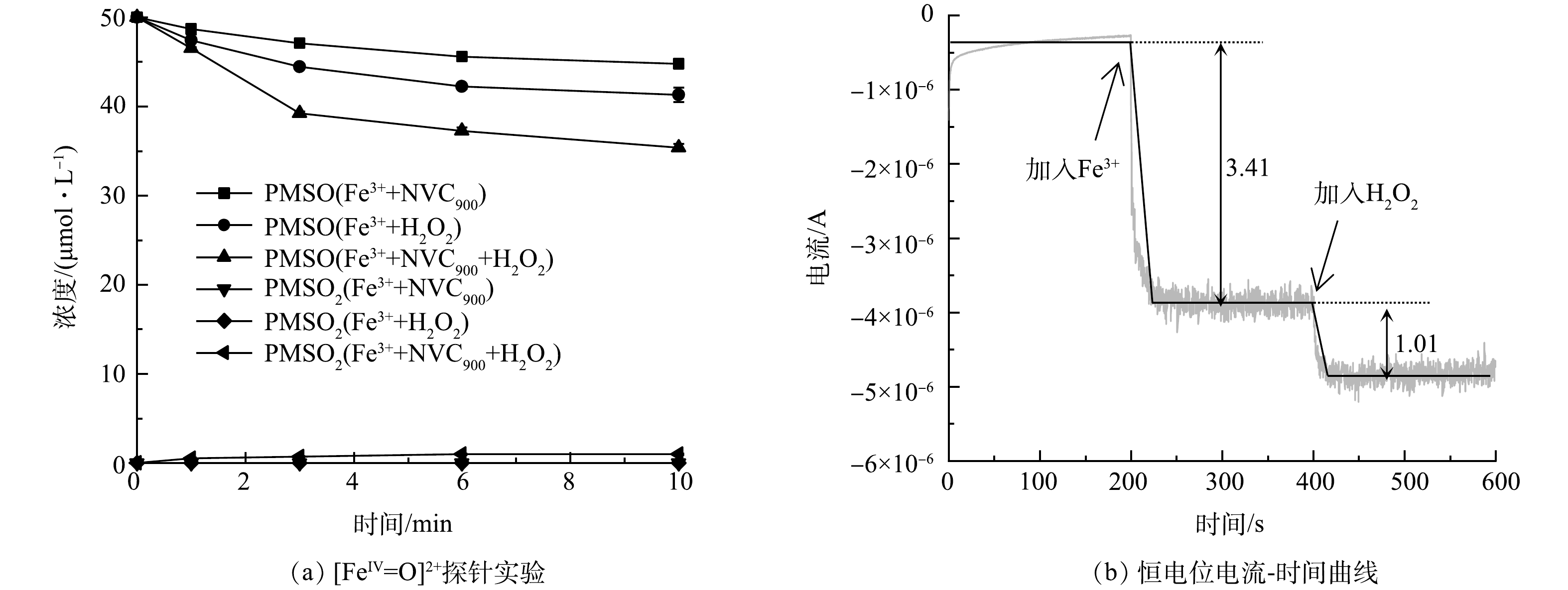
图 5 高价铁探针实验和电化学分析
Figure 5. High-valent iron probe experiment and electrochemical analysis
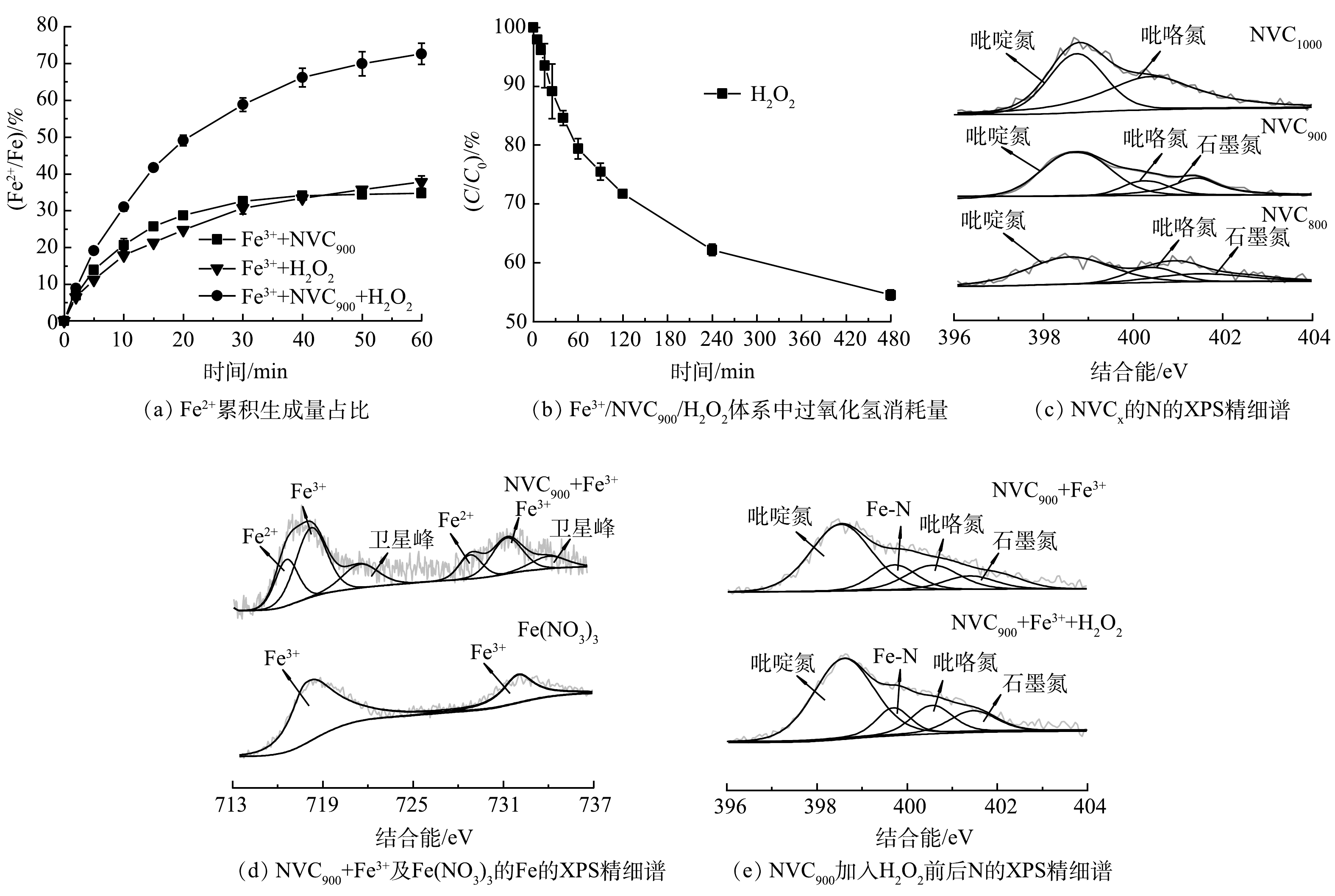
图 6 新生态Fe2+、Fe3+/NVC900/H2O2体系中H2O2消耗量和NVCx的助催化机制分析
Figure 6. Analysis of H2O2 consumption and NVCx-promoted catalytic mechanism in new ecological Fe2+、Fe3+/NVC900/H2O2 systems
-
[1] RAHMAN M S, ADEGOKE E O, PANG M-G. Drivers of owning more BPA[J]. Journal of Hazardous Materials, 2021, 417: 126076. doi: 10.1016/j.jhazmat.2021.126076 [2] XIAO C, WANG L, ZHOU Q, et al. Hazards of bisphenol A (BPA) exposure: A systematic review of plant toxicology studies[J]. Journal of Hazardous Materials, 2019, 384: 121488. [3] CIMMINO I, FIORY F, PERRUOLO G, et al. Potential mechanisms of bisphenol A (BPA) contributing to human disease[J]. International Journal of Molecular Sciences, 2020, 21(16): 5761. doi: 10.3390/ijms21165761 [4] 赵昌爽, 张建昆. 芬顿氧化技术在废水处理中的进展研究[J]. 环境科学与管理, 2014, 39(5): 83-87. doi: 10.3969/j.issn.1673-1212.2014.05.024 [5] 陆恬奕, 李宇, 徐瑞, 等. 高级氧化技术水处理研究进展[J]. 当代化工, 2021, 50(5): 1257-1260. [6] 孟琪莉, 孙冲. 高级氧化技术在工业难降解有机废水处理中的应用研究进展[J]. 工业用水与废水, 2021, 52(3): 1-5. [7] 李春娟. 芬顿法和类芬顿法对水中污染物的去除研究[D]; 哈尔滨: 哈尔滨工业大学, 2009. [8] MEKMOUCHE Y, MéNAGE S, TOIA-DUBOC C, et al. H2O2‐dependent Fe‐catalyzed oxidations: Control of the active species[J]. Angewandte Chemie, 2001, 113(5): 975-978. doi: 10.1002/1521-3757(20010302)113:5<975::AID-ANGE975>3.0.CO;2-P [9] CHEN L, MA J, LI X, et al. Strong enhancement on Fenton oxidation by addition of hydroxylamine to accelerate the ferric and ferrous iron cycles[J]. Environmental Science & Technology, 2011, 45(9): 3925-3930. [10] 李喜坤, 修稚萌, 孙旭东, 等. 碳热还原法制备 Ti (C, N) 粉末[J]. 粉末冶金工业, 2004, 14(1): 18-22. doi: 10.3969/j.issn.1006-6543.2004.01.004 [11] SUN D, SHEN B, YANG S, et al. Nitrogen-doped CNTs enhance heterogeneous Fenton reaction for IOH removal by FeOCl: Role of NCNTs and mechanism[J]. Separation and Purification Technology, 2023, 326: 124763. doi: 10.1016/j.seppur.2023.124763 [12] TROYANO J, CARNé-SáNCHEZ A, AVCI C, et al. Colloidal metal–organic framework particles: the pioneering case of ZIF-8[J]. Chemical Society Reviews, 2019, 48(23): 5534-5546. doi: 10.1039/C9CS00472F [13] DU P D, HIEU N T, THIEN T V. Ultrasound-assisted rapid ZIF-8 synthesis, porous ZnO preparation by heating ZIF-8, and their photocatalytic activity[J]. Journal of Nanomaterials, 2021, 2021: 9988998. [14] LONG X, LI Z, GAO G, et al. Graphitic phosphorus coordinated single Fe atoms for hydrogenative transformations[J]. Nature Communications, 2020, 11(1): 4074. doi: 10.1038/s41467-020-17903-0 [15] JIANG H-L, LIU B, LAN Y-Q, et al. From metal–organic framework to nanoporous carbon: toward a very high surface area and hydrogen uptake[J]. Journal of the American Chemical Society, 2011, 133(31): 11854-11857. doi: 10.1021/ja203184k [16] HU Y, KAZEMIAN H, ROHANI S, et al. In situ high pressure study of ZIF-8 by FTIR spectroscopy[J]. Chemical communications, 2011, 47(47): 12694-12696. doi: 10.1039/c1cc15525c [17] MA W, DU Y, WANG N, et al. ZIF-8 derived nitrogen-doped porous carbon as metal-free catalyst of peroxymonosulfate activation[J]. Environmental Science and Pollution Research, 2017, 24: 16276-16288. doi: 10.1007/s11356-017-9191-2 [18] LV C, QIAN Y, YAN C, et al. Defect engineering metal-free polymeric carbon nitride electrocatalyst for effective nitrogen fixation under ambient conditions[J]. Angewandte Chemie-International Edition, 2018, 57(32): 10246-10250. doi: 10.1002/anie.201806386 [19] RUSH J D, BIELSKI B H. Pulse radiolytic studies of the reaction of perhydroxyl/superoxide O2− with iron (II)/iron (III) ions. The reactivity of HO2/O2− with ferric ions and its implication on the occurrence of the Haber-Weiss reaction[J]. The Journal of Physical Chemistry, 1985, 89(23): 5062-5066. doi: 10.1021/j100269a035 [20] ANIPSITAKIS G P, DIONYSIOU D D, GONZALEZ M A. Cobalt-mediated activation of peroxymonosulfate and sulfate radical attack on phenolic compounds. Implications of chloride ions[J]. Environmental Science & Technology, 2006, 40(3): 1000-1007. [21] 许晟硕, 钱征, 王龄侦, 等. 氮掺杂碳催化剂活化过一硫酸盐的活性位点分析及其对双酚 A的降解机制[J]. 环境工程学报, 2022, 16(2): 452-461. doi: 10.12030/j.cjee.202111044 [22] DUAN X, SUN H, SHAO Z, et al. Nonradical reactions in environmental remediation processes: Uncertainty and challenges[J]. Applied Catalysis B: Environmental, 2018, 224: 973-982. doi: 10.1016/j.apcatb.2017.11.051 [23] YANG S, XU S, TONG J, et al. Overlooked role of nitrogen dopant in carbon catalysts for peroxymonosulfate activation: Intrinsic defects or extrinsic defects?[J]. Applied Catalysis B: Environmental, 2021, 295: 120291. doi: 10.1016/j.apcatb.2021.120291 [24] BUXTON G V, GREENSTOCK C L, HELMAN W P, et al. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−)in Aqueous Solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2): 513-886. doi: 10.1063/1.555805 [25] 张瑛洁, 马军, 张亮, 等. 树脂负载 FeⅢF催化过氧化氢降解染料孔雀石绿[J]. 环境科学学报, 2009(10): 2063-2069. doi: 10.3321/j.issn:0253-2468.2009.10.007 [26] 中华人民共和国生态环境部. 水质 铁的测定 邻菲啰啉分光光度法(试行): 标准号: HJ/T 345-2007[S]. 北京: 国家环境环保总局, 2007. [27] 陆平. 草酸钛钾分光光度法测定 Fenton 高级氧化系统中的过氧化氢[J]. 建筑工程技术与设计, 2014, 8: 582-,517. doi: 10.3969/j.issn.2095-6630.2014.36.257 [28] QIAN Z, WANG L, DZAKPASU M, et al. Spontaneous FeIII/FeII redox cycling in single-atom catalysts: Conjugation effect and electron delocalization[J]. iScience, 2023, 26(1): 105902. doi: 10.1016/j.isci.2022.105902 [29] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 16, Revision C. 01[M]. Wallin, 2016. [30] LU T, CHEN F. Multiwfn: A multifunctional wavefunction analyzer[J]. Journal of computational chemistry, 2012, 33(5): 580-592. doi: 10.1002/jcc.22885 [31] SONG X, SHI Y, WU Z, et al. Unraveling the discriminative mechanisms for peroxy activation via atomically dispersed Fe-N5 sites for tunable water decontamination[J]. Applied Catalysis B: Environmental, 2024, 340: 123240. doi: 10.1016/j.apcatb.2023.123240 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1528
- HTML全文浏览数: 1528
- PDF下载数: 47
- 施引文献: 0
