

 下载:
下载:
-
随着我国现代化的快速发展,伴生的各类污废水问题受到广泛的关注。各种新兴污染物的不断涌现,成为目前污水处理领域重点关注的问题,其中以酚类污染物最为典型。由于酚类污染物具有持久性,牵扯行业广泛,在地表水、地下水和农田灌溉水中均有相关检出的报道,已成为造成生态风险最为严重的污染物类型之一,对人类健康具有巨大的威胁[1-4]。芬顿反应由于其可产生强氧化性的活性物种(如羟基自由基、高价铁等),成为难降解有机废水,尤其是含酚类污染物废水的常用处理技术[5]。然而,芬顿氧化过程中存在Fe3+易水解失活、新生态Fe2+生成效率低下等关键问题,导致实际应用过程中存在反应pH条件敏感、投药量大、含铁污泥产量大等现实问题,导致其运行成本较高,严重阻碍了技术的广泛运用[6]。
针对上述问题,有相关研究在芬顿反应体系中引入乙二胺四乙酸(EDTA)、N,N'-(1,2-乙烷二基)双天冬氨酸(EDDS)等有机配体,通过配体与铁离子形成配合物,有效避免铁离子水解失活[7-8]。同时,形成的配合物可有效调控Fe3+/Fe2+循环过程的电极电位,缩小与H2O2之间的电位差,加速H2O2对Fe3+的还原过程,促进新生态Fe2+的形成。值得注意的是,许多单酚类污染物由于酚羟基的存在,同样具备与铁离子配位的能力[9]。邻苯二酚是造纸废水排放中的酚类污染物,具有显著的代表性[10-12]。有研究[13-15]表明,邻苯二酚可以和多种金属离子配位。使用芬顿氧化体系降解邻苯二酚类污染物时,污染物是否可对芬顿氧化体系产生与EDTA等有机配体类似的助催化作用从而加速自身的降解,此类研究报道较少。此外,含酚废水通常有复杂的共存有机污染物,基于以废治废的思路,邻苯二酚与芬顿氧化体系之间的协同作用对共存污染物的影响同样值得深入研究。
本研究构建了Fe3+/H2O2类芬顿高级氧化体系并将其用于降解水体中邻苯二酚,考查了反应条件对降解邻苯二酚效果的影响,通过顺磁共振、电子光谱、量子化学计算等手段对降解过程机制进行了详细分析,并对类芬顿氧化体系和邻苯二酚之间的协同作用进行了评估验证,探明了邻苯二酚对芬顿氧化体系的助催化特性。本研究结果可对缓解芬顿氧化应用过程中存在的关键问题可提供理论与数据参考。
-
邻苯二酚(C6H6O2)、硝酸铁(Fe(NO3)3·9H2O)、硝酸(HNO3)、氢氧化钠(NaOH)、叔丁醇(TBA)、卡马西平(C15H12N2O, carbamazepine, CBZ)、布洛芬(C13H18O2, ibuprofen, IBU)、阿特拉津(C8H14ClN5, atrazine, ATZ)、磺胺甲恶唑(C10H11N3O3S, sulfamethoxazole, SMX)、双酚A(C15H16O2, bisphenol A, BPA)等试剂均购自上海麦克林公司(分析纯),甲醇(色谱纯)、乙腈(色谱纯)等有机溶剂均购自赛默飞世尔科技公司,实验用水均出自Milli-Q超纯水系统。
-
1)实验仪器。高效液相色谱仪(Shimadzu LC-20A)用于降解实验中的数据测定;Bruker EMXmicro-6/1电子顺磁共振仪(EPR),用于活性物种识别;J6型紫外-可见光分光光度计用于测定反应体系吸收光谱;电化学工作站(CHI660E)用于体系反应过程中的电化学行为分析,采用三电极体系,以银-氯化银电极作为参比电极,铂电极作为对电极,玻碳电极作为工作电极,电解质采用0.1 mol·L−1硫酸钠溶液,实验中所有pH均采用STARTER3100高精度pH/电导率仪进行精确控制。
2)邻苯二酚降解实验。降解实验均在室温(25±0.5) ℃、120 mL烧杯中进行,具体步骤如下:向烧杯中加入一定量邻苯二酚储备液和硝酸铁储备液,加入超纯水稀释至100 mL并调节溶液pH至目标反应条件。加入一定量H2O2储备液触发氧化反应并开始计时,在反应特定时间取样,取样体积为1.0 mL,样品使用0.22 μm滤膜过滤,并用0.5 mL甲醇迅速淬灭,随后使用高效液相色谱仪对目标污染物浓度进行测定,所有批实验至少重复3次。测定过程流动相采用超声脱气后的2‰乙酸-纯水混合液和甲醇(体积比为50%:50%),流速为0.7 mL·min−1,检测波长为277 nm,柱温控制为35 ℃。
-
以反应温度为(25±0.5) ℃、邻苯二酚与Fe3+浓度为30 μmol·L−1、H2O2浓度为3.0 mmol·L−1、溶液初始pH=4.0等条件作为基准反应条件,考查了这些因素中单一因素变化对体系氧化性能的影响。首先,考察了不同初始pH条件下Fe3+/H2O2体系对邻苯二酚的降解性能。如图1(a)所示,在基准反应条件下,当溶液初始pH在2.0~6.0,Fe3+/H2O2体系均可在6 min内实现邻苯二酚的完全降解。当初始pH为4.0时,邻苯二酚的降解反应动力学常数达到了2.26 min−1。表明在弱酸性的反应条件下Fe3+/H2O2体系对邻苯二酚有极强的氧化性能。当体系初始pH为4.0且仅投加H2O2时作为对照,邻苯二酚的降解率仅为2.7%,表明H2O2自身难以实现对邻苯二酚的氧化[16]。但值得注意的是,仅有低浓度Fe3+(30 μmol·L−1)的投加且无H2O2共存的条件下,仍有约6%的邻苯二酚被降解。表明Fe3+与邻苯二酚共存时,邻苯二酚即可供出电子实现自身一定程度的降解。
其次,对Fe3+/H2O2体系降解邻苯二酚过程的最佳投药量进行了优化,结果如图1(b)所示。在基准反应条件下, H2O2浓度的变化对邻苯二酚的降解反应速率常数有较为显著的影响。其中,当H2O2浓度为1.0 mmol·L−1时,邻苯二酚的降解速率常数为1.85 min−1;当H2O2浓度增加至3.0 mmol·L−1时,降解速率常数达到2.26 min−1;当进一步增加H2O2投加量至5.0 mmol·L−1 (k=2.32 min−1)时,邻苯二酚降解的反应动力学常数并未随之继续升高,反而较投加量为4.0 mmol·L−1(k=2.44 min−1)时出现了轻微的降低,表明邻苯二酚的降解在较高H2O2浓度共存时受到一定的抑制。造成该结果的原因可能归因于以下2点:首先,过量H2O2的歧化反应(式(1))导致其无效分解;其次,过量的H2O2可对体系中的羟基自由基产生显著的淬灭效应(式(2)),从而导致污染物降解速率的降低。以上实验现象在以自由基为主要活性物种的高级氧化体系中较为常见[17],同时也说明羟基自由基是Fe3+/H2O2体系降解邻苯二酚过程中主要的活性物种。
在基准反应条件下,进一步考察了Fe3+浓度对Fe3+/H2O2体系降解邻苯二酚效果的影响,结果如图1(c)所示。可以看出,当Fe3+浓度在10.0~60.0 μmol·L−1内,邻苯二酚的降解速率随着Fe3+浓度的提高而升高,当Fe3+浓度增加至30.0 μmol·L−1时,邻苯二酚可在2 min内实现完全降解,高于10.0 μmol·L−1时的降解效率。最后,评估了常见共存离子对Fe3+/H2O2体系降解邻苯二酚过程的影响,结果见图1(d)。可见,当体系中存在30.0 μmol·L−1的CO32−、SO42−、Cl−、HCO3−和NO3−时,邻苯二酚均可在2 min内完全降解,表明水体中常见的共存离子对Fe3+/H2O2体系降解邻苯二酚的过程不产生显著影响,但在CO32−共存时体系污染物降解速率受到了轻微的抑制,可能是由于CO32−与羟基自由基发生反应,形成了氧化性能较弱的碳酸根自由基。
上述结果表明,Fe3+/H2O2高级氧化体系在复杂水质和较宽的pH范围内对邻苯二酚有较高的降解效率。但是,该体系仍然对溶液pH较为敏感,当pH接近中性时,体系的氧化性能难以保持。在基准反应条件下,Fe3+的投加浓度对邻苯二酚降解过程的影响最为显著。当Fe3+浓度达到60.0 μmol·L−1时,邻苯二酚可在1min内实现完全降解。最后,对基准反应条件下Fe3+/H2O2降解邻苯二酚过程的总有机碳去除率进行了分析,结果如图1(e)所示。当反应时间达到120 min时,TOC的去除率达到了43.67%,这表明邻苯二酚在Fe3+/H2O2体系氧化过程中可实现矿化。
-
活性物种是高级氧化技术实现难降解有机污染物降解的主要原因。为了解Fe3+/H2O2体系高效降解邻苯二酚过程的活性物种类型与产生机制,本研究进一步以5,5-二甲基-1-吡咯啉-N-氧化物(5,5-dimethyl-1-pyrroline N-oxide, DMPO)作为自由基捕获剂,通过EPR对反应过程中产生的活性物种进行了鉴定分析,结果如图2(a)所示。可以发现,在加入H2O2后,峰强比约为1:2:2:1的四线峰始终保持较大强度,此峰为典型的DMPO羟基加成产物(DMPO-OH),表明反应体系中产生了大量羟基自由基(HO·)。此外,在未加入H2O2的Fe3+与邻苯二酚的混合液中,可识别到一组较明显的六线峰,通过图谱指认,该六线峰常出现在烃基自由基存在的反应体系中。值得注意的是,当H2O2投加至反应体系后,该组六线峰信号持续存在,并与羟基自由基加成产物的信号共存,表明体系中存在类似烃基自由基的有机物中间体。对比无H2O2投加条件下Fe3+对邻苯二酚的直接降解图谱(图1(a)),表明Fe3+与邻苯二酚的共存直接导致邻苯二酚转化为含有类似烃基自由基的中间体,该中间体具有未成对的单电子结构,易被DMPO捕获形成对应的加成产物。换言之,邻苯二酚在与Fe3+共存的过程中供出了电子,该现象值得重点关注。
在甲醇溶液中以DMPO作为捕获剂对Fe3+/H2O2氧化体系降解邻苯二酚过程进行了顺磁共振(EPR)分析(图2(b)),在图谱中可观测到一类组六线峰,推测为典型的超氧化物加成产物DMPO-O2·−的特征峰。该结果表明体系中存在超氧自由基(O2·−),其可能的产生途径如式(3)~(4)[18]所示。图2(c)中给出了2,2,4,6-四甲基哌啶(2,2,4,6-tetramethylpiperidine, TEMP)作为自旋捕获剂的EPR分析结果。由图谱中可发现峰强比约为1:1:1的三线峰,对应TEMP-1O2吸收峰。此外,该三线峰的峰强随着反应时间的延长强度变化较小,表明1O2在Fe3+/H2O2降解邻苯二酚的体系中同样有显著的生成。根据前期研究结果[19],O2·−的歧化反应可能是体系中检测1O2信号的主要原因(式(5))。
通过EPR分析,确定了体系中产生的活性物种类型。为明确各活性物种对类芬顿氧化体系的贡献,选用叔丁醇(TBA, k(TBA, HO·)=6.0×108 mol·(L·s)−1, k(TBA, 1O2)=1.8×103 mol·(L·s)−1)、对苯醌(p-BQ, k(p-BQ, HO·)=1.2×109 mol·(L·s)−1, k(p-BQ, O2·−)=8.3×108 mol·(L·s)−1, k(p-BQ, 1O2)=6.6×107 mol·(L·s)−1)和糠醇(FFA, k(FFA, HO·)=1.5×1010 mol·(L·s)−1, k(FFA, O2·−)=3.5×103 mol·(L·s)−1, k(FFA, 1O2)=1.2×108 mol·(L·s)−1)作为淬灭剂,对反应体系进行了淬灭实验[20]。由图2(d)可知,各淬灭剂对体系的降解效果均有显著的抑制,由于上述淬灭剂均与HO·具有最高的反应速率,因此,可以推测出HO·是该体系中实现污染物降解的主要活性物种。有别于异相活化反应,对于均相活化反应而言,淬灭剂共存通常可完全掩蔽体系的氧化性能。在本研究中,TBA、p-BQ和FFA等均未完全中止体系对邻苯二酚的降解,表明体系中存在未知的邻苯二酚氧化路径,可能与EPR分析中烃基自由基的形成有直接的关系。
-
为探究淬灭剂无法完全中止均相活化反应的现象,本研究对Fe3+与邻苯二酚共存体系的电子光谱进行了详细分析,结果如图3(a)所示。可以发现,Fe3+溶液的吸收峰出现于292 nm,对应Fe3+的3d轨道内电子d-d跃迁。由于该跃迁禁阻,故以吸收带的形式呈现。相比之下,邻苯二酚溶液的吸收峰出现于274 nm,主要为邻苯二酚的分子内电荷迁移跃迁导致。分子内电荷跃迁主要有σ→σ*跃迁、n→σ*跃迁、π→π*跃迁和n→π*跃迁4种方式[21],由于邻苯二酚分子中氧原子的孤对电子和苯环上大π键的共轭效应,导致邻苯二酚具有大量的孤对电子与离域电子,因此,该峰被认为是n→π*跃迁或π→π*跃迁导致。需要重点注意的是,当Fe3+与邻苯二酚共存时,其电子光谱与Fe3+溶液和邻苯二酚溶液单独存在时完全不同。首先,在400 nm处与大于600 nm处,出现了2个宽吸收带,通常出现在具有裸眼视觉色配合物的电子光谱中。该现象进一步证明了Fe3+与邻苯二酚的共存直接形成了配合物(Fe3+-邻苯二酚),而上述2个宽吸收带可指认为该配合物的配位场光谱(即d-d跃迁光谱)或电荷迁移光谱。更重要的是,274 nm处原本为邻苯二酚分子内电子跃迁的峰也发生了约3 nm的蓝移,表明配合物中存在配体到金属的电荷迁移(ligand to metal charge transfer, LMCT)。LMCT过程的发现表明邻苯二酚中的大π键与铁的3d轨道形成了π型配键,在配位场的驱动下,通过电子离域过程实现了配体中π电子向Fe3+的供出,使其自身具有了有机自由基的特征。以上反应过程直接解释了烃基自由基的形成原因,也解释了无H2O2共存的反应条件下,Fe3+对邻苯二酚的轻微降解原理。
为深入理解Fe3+-邻苯二酚配合物的化学性质,本研究对不同pH条件下,Fe3+-邻苯二酚配合物的电子光谱进行了进一步分析(图3(b))。结果表明,Fe3+-邻苯二酚配合物的可见光吸收波段随着所处溶液pH的升高发生了显著的蓝移,该现象直接表明Fe3+与邻苯二酚形成配合物的LMCT过程随着pH发生了显著的变化,根据现有研究的结果[22],原因可能是溶液pH对配位状态的影响,通过该结果可以进行合理推测,在pH=6.0的条件下,Fe3+/H2O2体系对邻苯二酚应具有最强的降解效果。但是,该推论与邻苯二酚降解实验有明显的矛盾。在pH=6.0的情况下,邻苯二酚的降解效率受到了显著的抑制。为进一步分析该过程的反应机制,对不同体系中的再产生的Fe2+浓度进行了检测分析(图3(c-d),图中邻苯二酚标记为CTL)。相比pH=4.0时Fe3+/H2O2体系中极低的Fe2+浓度(0.26 μmol·L−1),Fe3+/邻苯二酚体系中产生了大量的Fe2+(7.26 μmol·L−1),表明LMCT可有效强化Fe3+/Fe2+循环。同时需要注意,Fe3+/H2O2/邻苯二酚体系中Fe2+浓度达到14.86 μmol·L−1,大于各反应试剂单独存在时产生Fe2+之和,表明邻苯二酚与H2O2协同配位可同时促进Fe价态循环和H2O2活化。随后检测了不同pH条件下Fe3+/邻苯二酚体系中的Fe2+浓度,结果表明当pH由2.0逐步升高至6.0时,Fe2+浓度由0.17 μmol·L−1升高至8.45 μmol·L−1,表明LMCT机制在较高的pH条件下由于邻苯二酚配位数的增加拥有更大的强度。因此,在pH=6.0条件下,大幅度降低的邻苯二酚降解效果可以被解释为在该条件下,Fe3+-邻苯二酚的配合物具有六配位的结构,H2O2无法直接接触到铁中心实现活化。而在pH相对温和的弱酸性和酸性条件下,Fe3+-邻苯二酚配合物可能以四配位或二配位的形式存在。H2O2可通过空余的配位数实现与铁中心的接触,从而通过配体至金属的电子迁移实现活化。可以合理推测的是,四配位状态下,配合物兼具空余的配位数和最丰富的离域电子,可实现H2O2的高效活化。
上述过程机制通过恒电位电流-时间曲线电化学分析进行了进一步验证。如图3(e)所示,向0.15 mmol·L−1的邻苯二酚溶液中施加0.294 V的恒电位时(由电化学开路电压(open-circuit potential, OCP)测得),测得稳定电流强度0.129×10−5 A,待电流稳定200 s后向溶液中加入0.15 mmol·L−1的Fe(NO3)3溶液,电流强度迅速波动下降并缓慢稳定于−3.40×10−5 A。结果表明邻苯二酚向Fe3+传递电荷,形成Fe3+-邻苯二酚配合物。进一步向体系中添加15 mmol·L−1 H2O2后电流下降并稳定于−3.63×10−5 A。表明Fe3+-邻苯二酚配合物的LMCT机制可有效活化H2O2,结合活性物种分析研究(图2),相关过程如式(6)~(8)所示,式中邻苯二酚简写为CTL。
-
最后,本研究采用Gaussian 16 W C.01量子化学软件对Fe3+/H2O2氧化体系降解邻苯二酚过程的详细机制进行了分析。使用密度泛函理论(density functional theory, DFT)计算[23],计算泛函采用B3LYP方法,采用6−31+G(d, p)全电子基组[24-25]。
首先对二配位、四配位和六配位形式的Fe3+-邻苯二酚进行了几何结构优化,并在优化后的构型基础上进行能量计算,几何优化后的构型结果见图4,分子团簇中各原子Mulliken电荷变化情况见图4,可以发现,当Fe3+与邻苯二酚分别以二、四、六配位的方式形成配合物并达到热力学稳定状态后,铁中心的Mulliken电荷各自由+3降低至+1.018、+0.810和+0.479。表明随着与Fe3+配位的邻苯二酚数量的增加,配合物团簇中铁中心的还原性持续增强。该结果与Fe2+定量分析的结论完全一致,直接证明了LMCT在配合物中的重要作用。此外,对四配位状态下H2O2接触金属中心的过程进行了计算分析。由图4可见,当H2O2通过空余配位数与铁中心配位后,铁中心的Mulliken电荷由+0.810上升至+0.845。同时,配位态H2O2的Mulliken电荷由0降低至−0.612。此外,O-O键键长由0.148 nm伸长至0.264 nm,表明Fe3+/邻苯二酚通过铁中心将电子传递给H2O2,使得其脱离基态并导致H2O2伸缩振动模式发生显著变化,随后实现其活化并释放活性物种。
-
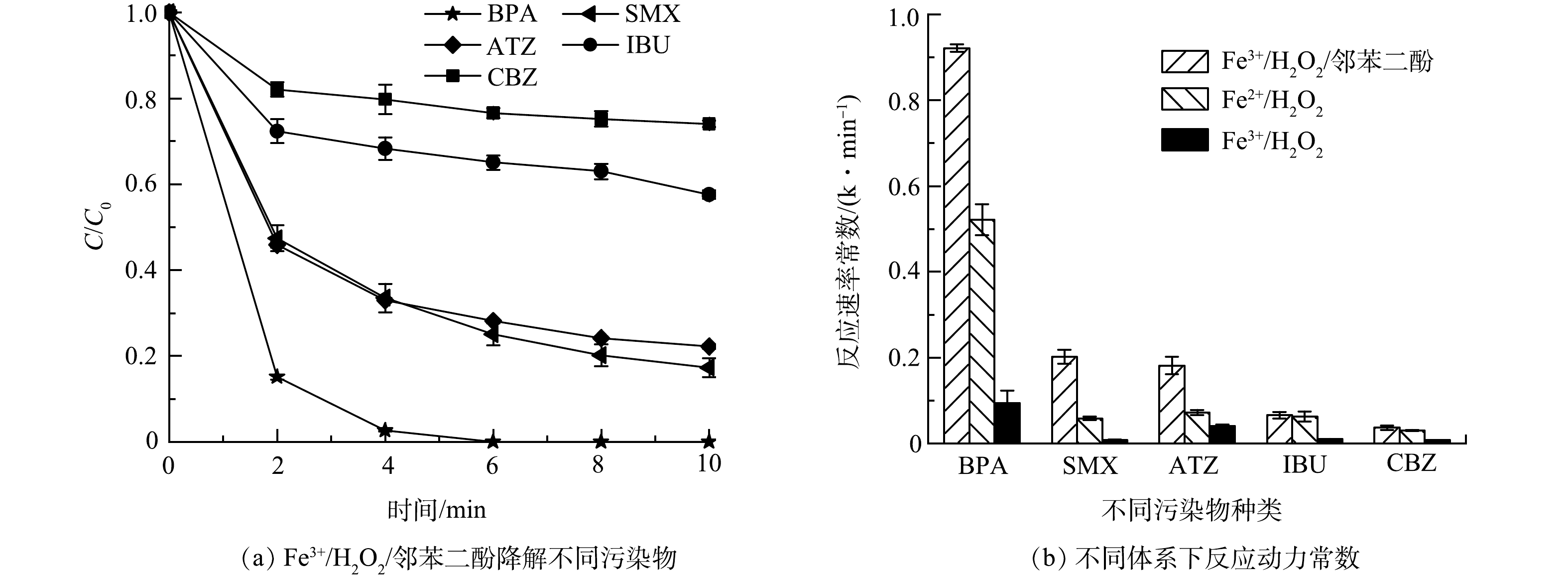
根据机理分析结果,Fe3+/H2O2体系对邻苯二酚的降解由自由基氧化和LMCT过程共同作用导致。由于LMCT过程的存在,邻苯二酚与Fe3+配位后,可有效的实现自身电子向中心铁原子的迁移。进而促进了Fe3+/Fe2+的循环,使其更易实现共存H2O2的活化,实现活性物种的高效释放。因此,邻苯二酚在与Fe3+配位的过程中自发的起到了助催化剂的作用,有效避免了芬顿氧化技术中存在的Fe3+/Fe2+的循环效率低的关键问题,强化了其自身的降解。由于在邻苯二酚的废水中同时存在其他有机污染物,因此,对共存有机物的降解性能值得深入评估。如图5(a)与图5(b)所示,当反应温度为(25±0.5) ℃、邻苯二酚与Fe3+浓度均为30 μmol·L−1、溶液pH=4.0,H2O2投加浓度为3.0 mmol·L−1时,对体系中共存的新污染物的降解性能进行了分析(新污染物共存浓度均为50 μmol·L−1)。可以看出,Fe3+/H2O2/邻苯二酚三元体系可有效地降解共存有机污染物。在10 min内,对BPA、SMX、ATZ、IBU和CBZ的降解率分别达到了100%、82.73%、77.84%、42.40%和25.94%,对应降解动力学常数为0.92、0.20、0.18、0.07和0.04 min−1。在相同反应条件下,Fe3+/H2O2体系对上述污染物的降解反应速率极低。更重要的是,BPA、SMX、ATZ、IBU和CBZ在芬顿氧化体系(Fe2+/H2O2)中的降解反应速率常数分别为0.52、0.06、0.07、0.06和0.03 min−1,同样显著低于Fe3+/H2O2/邻苯二酚体系中的降解速率。传统芬顿氧化体系为了抑制Fe3+的水解,通常需要保持较低的pH环境,并通过H2O2对Fe3+实现缓慢的还原。邻苯二酚的共存有效避免了Fe3+的水解,并强化了Fe2+的再生。因此,在相同pH条件下,Fe3+/H2O2/邻苯二酚体系对共存有机污染物展现出了高于芬顿氧化体系的降解效率。由此可见,作为典型新污染物的邻苯二酚,与Fe3+/H2O2体系的共存,不仅可以有效的实现自身的降解,还可以实现对体系中共存有机污染物的高效降解。
-
本文探究了Fe3+/H2O2体系降解邻苯二酚的最佳反应条件及高级氧化途径,对其中LMCT机制强化Fe3+/Fe2+循环,进而提升Fe3+/H2O2氧化能力的过程机理进行了详细分析。通过对多类型共存有机污染物的降解实验,分析了邻苯二酚作为芬顿氧化体系助催化剂的特性,主要结论如下。
1)在反应温度为(25±0.5) ℃、Fe3+浓度为30 μmol·L−1、初始pH=4.0,H2O2投加量3.0 mmol·L−1的条件下,Fe3+/H2O2体系对邻苯二酚达到了最佳的降解速率,该过程以H2O2活化产生的HO·为主导活性物种实现。
2)邻苯二酚与Fe3+的共存可形成配位物,有效地缓解了芬顿氧化体系中存在的Fe3+易水解的关键问题,此外,配合物中配体到铁中心的电荷迁移强化了新生态Fe2+的生成,强化了对H2O2的活化行为。
3)配体到铁中心的电荷迁移过程不仅有利于邻苯二酚自身的降解,并可使邻苯二酚产生助催化剂的作用,从而对CBZ、IBU、ATZ、SMX和BPA等共存有机物的降解产生显著的促进作用,且污染物降解速率显著高于传统芬顿氧化体系。
Fe3+/H2O2氧化体系降解邻苯二酚的过程机制
Process mechanism on catechol degradation by Fe3+/H2O2 oxidation system
-
摘要: 本研究构建了Fe3+/H2O2氧化体系,且对其降解水中邻苯二酚的效果进行了研究,考察了不同反应条件对体系降解性能的影响,借助顺磁共振、活性物种淬灭、量子化学等手段对降解过程机制进行了详细分析。结果表明,Fe3+/H2O2氧化体系可以在弱酸条件下实现邻苯二酚的高效降解。顺磁共振分析与淬灭实验结果表明,降解过程除羟基自由基的生成外,体系中共存有大量烃基自由基。结合电子光谱分析结果发现,邻苯二酚在被降解的过程中与Fe3+形成了弱场配合物,该配合物不仅可在较温和的pH条件下实现Fe3+的水解抑制,还可通过配体到Fe3+的电荷迁移促进Fe(II)-Fe(III)-Fe(II)的循环,从而加速自身的降解。更重要的是,Fe3+/H2O2/邻苯二酚氧化体系可对水中共存的其他有机污染物产生显著的降解效果。以上研究结果可为抑制芬顿氧化过程中铁离子易水解和价态循环效率低的难题提供新的思路。Abstract: Fe3+/H2O2 oxidation system was developed in this study to explore catechol degradation effect in water. The oxidation performances of this system under different reaction conditions were examined, its mechanism was investigated through electron paramagnetic resonance (EPR), reactive oxidation species (ROS) scavenging experiment, and quantum chemistry theoretical calculation. The results showed that catechol could be degraded by Fe3+/H2O2 system efficiently under mild acidic conditions, EPR and scavenging tests results indicated that large amounts of alkyl radicals besides hydroxyl radicals were produced. Electron spectroscopy analysis revealed a weak-field coordination complex occurred between catechol and Fe3+ during degradation, which could inhibit Fe3+ hydrolysis under mild pH conditions and further promote Fe(II)-Fe(III)-Fe(II) redox cycling through charge transfer from ligand to metal, and thereby accelerating its self-degradation. Fe3+/H2O2 oxidation system could significantly enhance the degradation efficiency of coexisting organic compounds. This work provides a novel approach to face facile hydrolysis of iron and low redox cycling in Fenton process.
-
Key words:
- catechol /
- Fenton reaction /
- catalysis promoter /
- coordination chemistry /
- reactive oxidation species
-

-

图 1 Fe3+/H2O2体系降解邻苯二酚的性能与影响因素分析
Figure 1. Performance and influencing factors analysis of catechol degradation by Fe3+/H2O2 system
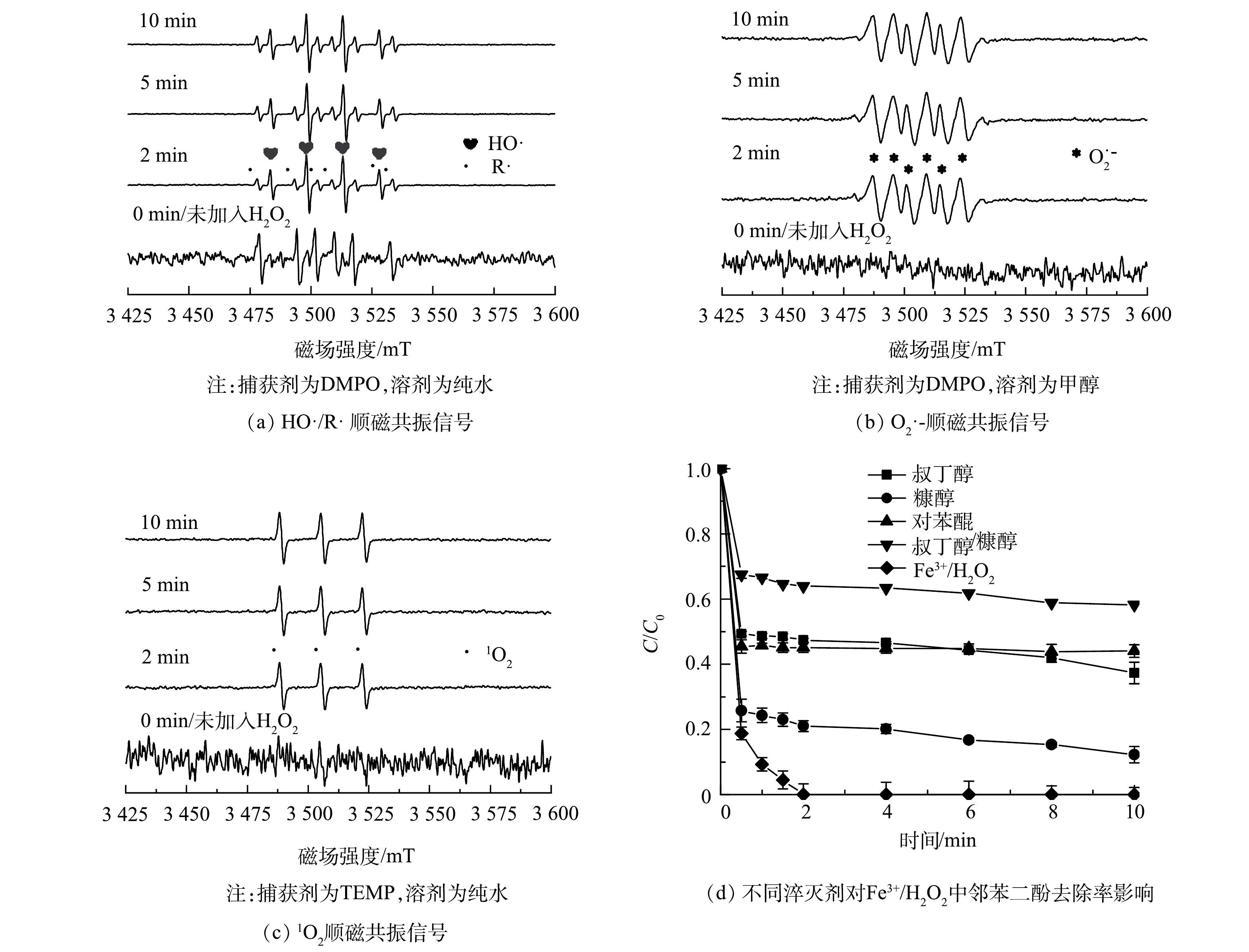
图 2 活性物种识别与淬灭实验
Figure 2. Identification of different reactive oxidation species and their scavenging experiments
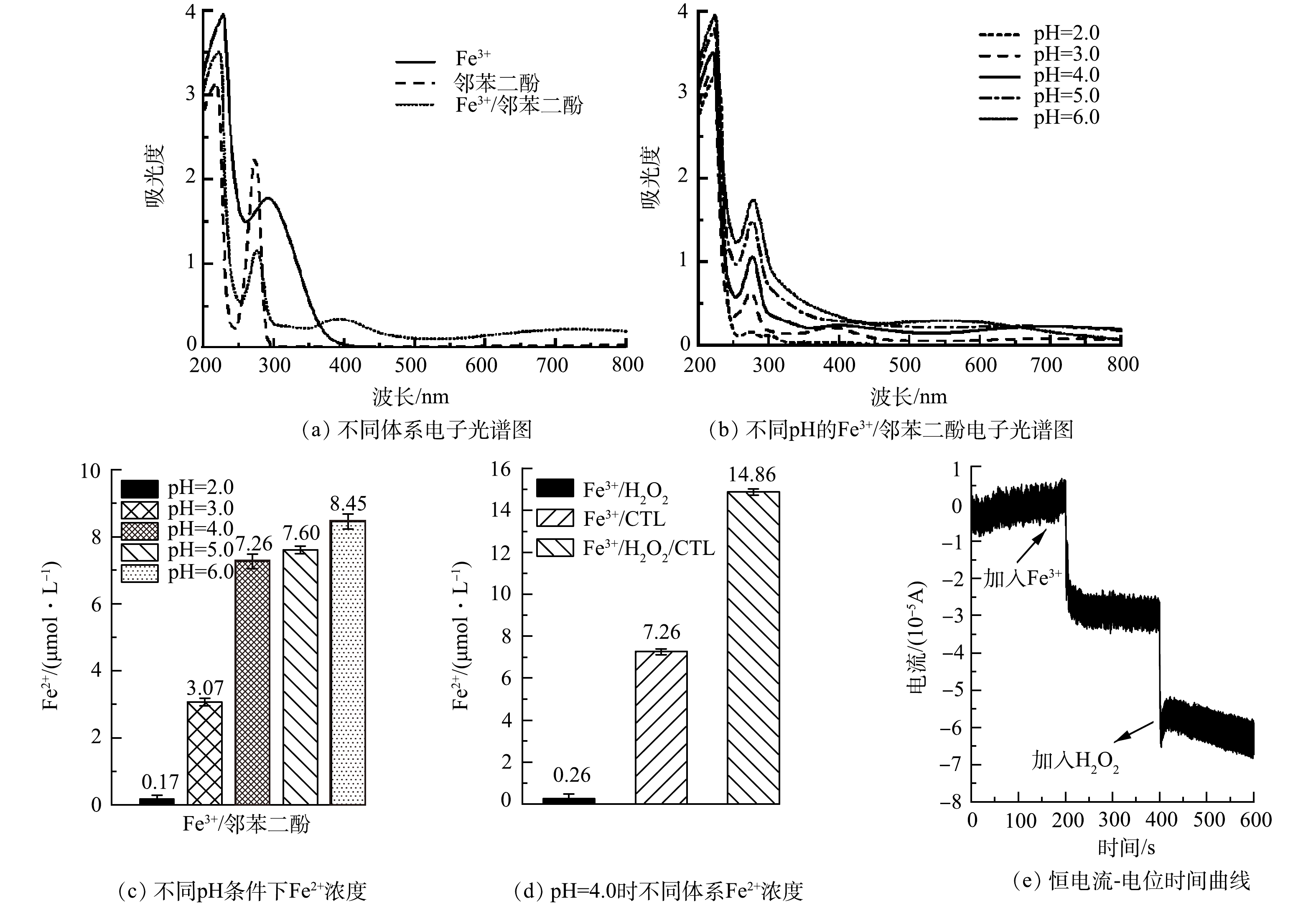
图 3 邻苯二酚/Fe3+体系中配体到金属的电荷迁移机制分析
Figure 3. Analysis of charge transfer mechanism from ligand to metal in the system of catechol/Fe3+

图 4 Fe3+/H2O2降解邻苯二酚过程的密度泛函理论计算分析
Figure 4. Density functional theory calculation analysis of catechol degradation in Fe3+/H2O2 process
-
[1] POZA-NOGUEIRAS V, ROSALES E, PAZOS M, et al. Current advances and trends in electro-Fenton process using heterogeneous catalysts: A review[J]. Chemosphere, 2018, 201: 399-416. doi: 10.1016/j.chemosphere.2018.03.002 [2] SAPUTERA W H, PUTRIE A S, ESMAILPOUR A A, et al. Technology advances in phenol removals: Current progress and future perspectives[J]. Catalysts, 2021, 11(8): 998. doi: 10.3390/catal11080998 [3] XIA C, LI X, WU Y, et al. A review on pollutants remediation competence of nanocomposites on contaminated water[J]. Environmental Research, 2023, 222: 115318. doi: 10.1016/j.envres.2023.115318 [4] YAO L, YANG H, CHEN Z, et al. Bismuth oxychloride-based materials for the removal of organic pollutants in wastewater[J]. Chemosphere, 2021, 273: 128576. doi: 10.1016/j.chemosphere.2020.128576 [5] 孟琪莉, 孙冲. 高级氧化技术在工业难降解有机废水处理中的应用研究进展[J]. 工业用水与废水, 2021, 52(3): 1-5. [6] CHEN L W, MA J, LI X C, et al. Strong enhancement on Fenton oxidation by addition of hydroxylamine to accelerate the ferric and ferrous iron cycles[J]. Environmental Science & Technology, 2011, 45(9): 3925-3930. [7] ZHANG L, WU B, ZHANG G, et al. Enhanced decomplexation of Cu(II)-EDTA: The role of acetylacetone in Cu-mediated photo-Fenton reactions[J]. Chemical Engineering Journal, 2019, 358: 1218-1226. doi: 10.1016/j.cej.2018.10.124 [8] YE Z, BRILLAS E, CENTELLAS F, et al. Expanding the application of photoelectro-Fenton treatment to urban wastewater using the Fe(III)-EDDS complex[J]. Water Research, 2020, 169: 115219. doi: 10.1016/j.watres.2019.115219 [9] LI X, PARK I, TABELIN C B, et al. Enhanced pyrite passivation by carrier-microencapsulation using Fe-catechol and Ti-catechol complexes[J]. Journal of Hazardous Materia, 2021, 416: 126089. doi: 10.1016/j.jhazmat.2021.126089 [10] 刘亦珍. 含酚废水治理技术研究现状及其进展[J]. 资源节约与环保, 2019(12): 91. doi: 10.3969/j.issn.1673-2251.2019.12.082 [11] 孙海丽, 李春琴, 岳斌, 等. 苯酚废水处理研究进展[J]. 绿色科技, 2021, 23(06): 56-57. doi: 10.3969/j.issn.1674-9944.2021.06.018 [12] 张国卿 王, 徐高田, 甘晓明. Fenton试剂在处理难降解有机废水中的应用[J]. 工业安全与环保, 2004(3): 17-30. [13] M'HEMDI A, DBIRA B, ABDELHEDI R, et al. Mineralization of catechol by Fenton and photo-Fenton processes[J]. Clean-Soil Air Water, 2012, 40(8): 878-885. doi: 10.1002/clen.201100376 [14] Cao D, Sun L, Wang G, et al. Kinetics of hydrogen peroxide electroreduction on Pd nanoparticles in acidic medium[J]. Journal of Electroanalytical Chemistry, 2008, 621(1): 31-37. doi: 10.1016/j.jelechem.2008.04.007 [15] XIAO J, WANG C, LYU S, et al. Enhancement of Fenton degradation by catechol in a wide initial pH range[J]. Separation and Purification Technology, 2016, 169: 202-209. doi: 10.1016/j.seppur.2016.04.031 [16] WANG Y, YE D, XU Y, et al. An electrochemical paper-based analytical device with facile carbon fiber-sewed electrodes for highly sensitive detection of hydrogen peroxide in real water[J]. Electrochimica Acta, 2024: 144091. [17] IBRAHIM M E A, NOME R A. Hydrogen peroxide disproportionation: time-resolved optical measurements of spectra, scattering and imaging combined with correlation analysis and simulations[J]. Journal of Molecular Structure, 2022, 1251: 131992. doi: 10.1016/j.molstruc.2021.131992 [18] ZHANG C, LI T, ZHANG J, et al. Degradation of p-nitrophenol using a ferrous-tripolyphosphate complex in the presence of oxygen: The key role of superoxide radicals[J]. Applied Catalysis B: Environmental, 2019, 259: 118030. doi: 10.1016/j.apcatb.2019.118030 [19] 许晟硕, 钱征, 王龄侦, 等. 氮掺杂碳催化剂活化过一硫酸盐的活性位点分析及其对双酚A的降解机制[J]. 环境工程学报, 2022, 16(2): 452-461. doi: 10.12030/j.cjee.202111044 [20] GUO Y, LONG J, HUANG J, et al. Can the commonly used quenching method really evaluate the role of reactive oxygen species in pollutant abatement during catalytic ozonation?[J]. Water Research, 2022, 215: 118275. doi: 10.1016/j.watres.2022.118275 [21] YANG, YANG, MARK, et al. Multireference Ab initio study of ligand field d–d transitions in octahedral transition-metal oxide clusters[J]. The Journal of Physical Chemistry C, 2014, 118(50): 29196-29208. doi: 10.1021/jp5052672 [22] HIROTAKA E, JOSEPH J, RICHARDSON, et al. One-step assembly of coordination complexes for versatile film and particle engineering[J]. Science, 2013, 6142: 154-157. [23] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 16, Revision C. 01[M]. Wallingford, CT. Delaware: Gauss instruments inc, 2016. [24] LU T, CHEN F W. Multiwfn: A multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 2012, 33(5): 580-592. doi: 10.1002/jcc.22885 [25] LU T, CHEN Q X. A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure[J]. Theoretical Chemistry Accounts, 2020, 139(2): 25-37. doi: 10.1007/s00214-019-2541-z -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1350
- HTML全文浏览数: 1350
- PDF下载数: 48
- 施引文献: 0
