

































































Concentration </td><td class="table_top_border" style="height:20pt;" colspan="2" align="center" valign="middle">HO<sup>•</sup></td><td class="table_top_border" style="height:20pt;" align="center" valign="middle"></td><td class="table_top_border" style="height:20pt;" colspan="2" align="center" valign="middle"><inline-formula><tex-math id="Z-20220127094341">${\rm{SO}}_4^{ \cdot - } $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" style="height:20pt;" rowspan="2" align="center" valign="middle">目标物Reactive species</td><td class="table_top_border" style="height:20pt;" rowspan="2" align="center" valign="middle">浓度/(mol·L<sup>−1</sup>)Concentration </td><td class="table_top_border" style="height:20pt;" colspan="2" align="center" valign="middle">HO<sup>•</sup></td><td class="table_top_border" style="height:20pt;" align="center" valign="middle"></td><td class="table_top_border" style="height:20pt;" colspan="2" align="center" valign="middle"><inline-formula><tex-math id="Z-20220127094341">${\rm{SO}}_4^{ \cdot - } $</tex-math><alternatives><img class="graphic" src="2020093004_Z-20220127094341.jpg"><img class="graphic" src="2020093004_Z-20220127094341.png"></alternatives></inline-formula></td></tr><tr> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>c</i>/ (L·mol<sup>−1</sup>·s<sup>−1</sup>)</td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>ck</i> /s<sup>−1</sup></td><td align="center" valign="middle" style="height:20pt;"></td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>k</i> /(L·mol<sup>−1</sup>·s<sup>−1</sup>)</td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>ck</i> /s<sup>−1</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:20pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">1.0 × 10<sup>−5</sup></td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">7.5 × 10<sup>9</sup></td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">7.5 × 10<sup>4</sup></td> <td class="table_bottom_border table_top_border2" style="width:0.5%;height:20pt;" align="center" valign="middle" rowspan="6"> </td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle">9.2 × 10<sup>9</sup></td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">9.2 × 10<sup>4</sup></td> </tr><tr><td rowspan="3" align="center" valign="middle" style="height:20pt;">TBA</td> <td align="center" valign="middle" style="height:20pt;">1.0 × 10<sup>−2</sup></td> <td rowspan="3" align="center" valign="middle" style="height:20pt;">7.6 × 10<sup>8</sup></td> <td align="center" valign="middle" style="height:20pt;">7.6 × 10<sup>6</sup></td> <td rowspan="3" align="center" valign="middle" style="height:20pt;">4.0 × 10<sup>5</sup></td> <td align="center" valign="middle" style="height:20pt;">4.0 × 10<sup>3</sup></td> </tr><tr> <td align="center" valign="middle" style="height:20pt;">1.0 × 10<sup>−1</sup></td> <td align="center" valign="middle" style="height:20pt;">7.6 × 10<sup>7</sup></td> <td align="center" valign="middle" style="height:20pt;">4.0 × 10<sup>4</sup></td> </tr><tr> <td align="center" valign="middle" style="height:20pt;">0.5</td> <td align="center" valign="middle" style="height:20pt;">3.8 × 10<sup>8</sup></td> <td align="center" valign="middle" style="height:20pt;">2.0 × 10<sup>5</sup></td> </tr><tr><td rowspan="2" align="center" valign="middle" class="table_bottom_border" style="height:20pt;">MeOH</td> <td align="center" valign="middle" style="height:20pt;">1.0 × 10<sup>−1</sup></td> <td rowspan="2" align="center" valign="middle" class="table_bottom_border" style="height:20pt;">9.7 × 10<sup>8</sup></td> <td align="center" valign="middle" style="height:20pt;">9.7 × 10<sup>7</sup></td> <td rowspan="2" align="center" valign="middle" class="table_bottom_border" style="height:20pt;">2.5 × 10<sup>7</sup></td> <td align="center" valign="middle" style="height:20pt;">2.5 × 10<sup>6</sup></td> </tr><tr> <td class="table_bottom_border" style="height:20pt;" align="center" valign="middle">0.5</td> <td class="table_bottom_border" style="height:20pt;" align="center" valign="middle">4.8 × 10<sup>8</sup></td> <td class="table_bottom_border" style="height:20pt;" align="center" valign="middle">1.3 × 10<sup>7</sup></td> </tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td></tr><tr> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>c</i>/ (L·mol<sup>−1</sup>·s<sup>−1</sup>)</td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>ck</i> /s<sup>−1</sup></td><td align="center" valign="middle" style="height:20pt;"></td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>k</i> /(L·mol<sup>−1</sup>·s<sup>−1</sup>)</td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle"><i>ck</i> /s<sup>−1</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:20pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">1.0 × 10<sup>−5</sup></td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">7.5 × 10<sup>9</sup></td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">7.5 × 10<sup>4</sup></td> <td class="table_bottom_border table_top_border2" style="width:0.5%;height:20pt;" align="center" valign="middle" rowspan="6"> </td><td class="table_top_border2" style="height:20pt;" align="center" valign="middle">9.2 × 10<sup>9</sup></td> <td class="table_top_border2" style="height:20pt;" align="center" valign="middle">9.2 × 10<sup>4</sup></td> </tr><tr><td rowspan="3" align="center" valign="middle" style="height:20pt;">TBA</td> <td align="center" valign="middle" style="height:20pt;">1.0 × 10<sup>−2</sup></td> <td rowspan="3" align="center" valign="middle" style="height:20pt;">7.6 × 10<sup>8</sup></td> <td align="center" valign="middle" style="height:20pt;">7.6 × 10<sup>6</sup></td> <td rowspan="3" align="center" valign="middle" style="height:20pt;">4.0 × 10<sup>5</sup></td> <td align="center" valign="middle" style="height:20pt;">4.0 × 10<sup>3</sup></td> </tr><tr> <td align="center" valign="middle" style="height:20pt;">1.0 × 10<sup>−1</sup></td> <td align="center" valign="middle" style="height:20pt;">7.6 × 10<sup>7</sup></td> <td align="center" valign="middle" style="height:20pt;">4.0 × 10<sup>4</sup></td> </tr><tr> <td align="center" valign="middle" style="height:20pt;">0.5</td> <td align="center" valign="middle" style="height:20pt;">3.8 × 10<sup>8</sup></td> <td align="center" valign="middle" style="height:20pt;">2.0 × 10<sup>5</sup></td> </tr><tr><td rowspan="2" align="center" valign="middle" class="table_bottom_border" style="height:20pt;">MeOH</td> <td align="center" valign="middle" style="height:20pt;">1.0 × 10<sup>−1</sup></td> <td rowspan="2" align="center" valign="middle" class="table_bottom_border" style="height:20pt;">9.7 × 10<sup>8</sup></td> <td align="center" valign="middle" style="height:20pt;">9.7 × 10<sup>7</sup></td> <td rowspan="2" align="center" valign="middle" class="table_bottom_border" style="height:20pt;">2.5 × 10<sup>7</sup></td> <td align="center" valign="middle" style="height:20pt;">2.5 × 10<sup>6</sup></td> </tr><tr> <td class="table_bottom_border" style="height:20pt;" align="center" valign="middle">0.5</td> <td class="table_bottom_border" style="height:20pt;" align="center" valign="middle">4.8 × 10<sup>8</sup></td> <td class="table_bottom_border" style="height:20pt;" align="center" valign="middle">1.3 × 10<sup>7</sup></td> </tr></tbody>
</table></div></foreignObject></svg>)













-
药物和个人护理产品(PPCPs)是一类 “新兴”环境污染物,它们具有在低剂量下诱导生物体产生生理效应的能力,作为医用或保健药品被广泛应用于人或动物[1]。然而,这些药物被人体或动物摄入后,仅少部分发生代谢,约30%—90%以原形药物排出体外,并不断释放到水环境中[2]。作为典型的PPCPs,双氯芬酸钠(diclofenac,DCF)是全球目前最畅销的解热镇痛消炎药物之一,每天约被世界各地3000万人广泛使用,目前已在各水环境中检出,如:污水处理厂、地表水、地下水、甚至自来水中,浓度在ng·L−1 — μg·L−1之间[3-4]。研究表明,痕量的DCF便可引起水生生物内脏器官发生病变[5-6],并通过食物链积累对生态系统和人类健康构成潜在威胁[7],其带来的环境污染问题也受到国内外广泛关注[8-10]。与大多数PPCPs相同,DCF难以被生物降解[11-12];目前,国内外主要采用化学氧化法(Fenton氧化法[13]、光化学氧化法[14-16]、电化学氧化法[17]、臭氧氧化法[18]、超声波辐射法[19-20]和光子束流辐射[21])对其降解。
近年来基于亚硫酸盐(S(Ⅳ))产生硫酸根自由基的高级氧化技术受到环境工作者关注,但主要用于大气治理和烟气脱硫,在水处理领域的应用研究仍处于起步阶段。从经济和毒性角度上看,亚硫酸盐较过硫酸盐具有价格低廉、半致死浓度更高等优点。目前,活化S(Ⅳ) 产生硫氧根自由基的方法主要有:UV辐射活化[22-23]和过渡金属活化[24-26]等。鉴于,Fe(Ⅲ)自然界中含量广泛,价格便宜、环境友好等特点,Fe(Ⅲ)活化S(Ⅳ)降解有机物具有应用及推广潜力。Yuan等[27]采用Fe(Ⅲ)/S(Ⅳ)降解苯胺,研究了不同浓度下反应剂对苯胺降解的影响;Zhou等[28]研究了Fe(Ⅲ)/S(Ⅳ)体系中不同硫氧自由基与酸性橙7的二级反应速率常数。现该技术的关注点主要为该体系的最佳反应条件及体系中存在的活性自由基,目标污染物的选取主要为有机染料、苯胺类有机物以及重金属等[29-30]。然而,将Fe(Ⅲ)/S(Ⅳ)作为高级氧化体系降解PPCPs类新兴有机物的研究报道较少。
本研究选用Fe(Ⅲ)活化亚硫酸氢钠(BS)处理DCF模拟废水,除了分析反应试剂对其降解的影响,还考察了水中常见干扰离子氯离子及黄腐酸(FA)的影响;并基于温度的影响计算了Fe(Ⅲ)/S(Ⅳ)体系的表观反应活化能;通过自由基抑制实验分析体系中的活性自由基;最后利用LC_MC测定DCF的降解中间产物,推测其可能的降解机理。
-
双氯芬酸钠(C14H10Cl2NO2Na,纯度≥98%)、1,10-菲罗啉(分析纯)、2-硝基苯甲酸和黄腐酸(FA,纯度≥90%)购于Aladdin公司;亚硫酸氢钠(NaHSO3)、九水硝酸铁(Fe(NO3)3·9H2O)、磷酸二氢钾(KH2PO4)、磷酸氢二钠(Na2HPO4)、氢氧化钠(NaOH)、硫代硫酸钠(Na2S2O3)、氯化钠(NaCl)、硫酸(H2SO4)和无水乙酸钠(CH3COONa)为分析纯,冰乙酸和叔丁醇为色谱纯,上述药品购于成都科龙化学品有限公司;甲醇(色谱纯)购于Fisher Scientific公司。去离子水(18MΩ·cm)用于试剂的溶解和配置。
-
实验在250 mL烧杯中进行,通过水浴恒温磁力搅拌装置来控制搅拌速度和反应温度,其中转子转速为600 r·min−1,温度设置为25 ℃(温度影响除外)。配置100 mL一定浓度的DCF溶液,投加一定量Fe(NO3)3储备溶液,然后用0.2 mol·L−1 NaOH和0.2 mol·L−1 H2SO4溶液将其pH值调节至预设值附近,投加1 mL NaHSO3储备溶液启动反应,反应过程中不对溶液pH进行控制。分别在0、0.5、1、2、3、5 min取1 mL样品放入装有50 μL的0.4 mol硫代硫酸钠的液相小瓶,快速摇匀终止反应后待测。所有实验至少进行两次,取平均值计算去除率。为确保加入NaHSO3后反应初始pH在设定值± 0.05范围内,需在投加Fe(NO3)3溶液后对pH进行多次微调和投加NaHSO3溶液预实验来确定预设值[31],除pH影响实验外,初始反应pH值均为4.0。除产物检测,所有实验至少进行2次,取平均值计算去除率。
-
DCF浓度测定采用高效液相色谱仪(Waters 2695)。具体参数:固定相为C18柱(5 μm × 4.6 mm × 150 mm);流动相由1‰乙酸水溶液与甲醇(25:75,V/V)组成,流速为1 mL·min−1;检测波长276 nm;进样体积为20 μL;柱温为30 ℃。
DCF中间产物的检测采用UPLC-QTOT-MS(Waters)。具体参数:色谱柱为BEH C18(1.7 μm × 2.1 mm × 50 mm);流动相为乙腈和水(0.1%甲酸),采用梯度洗脱的方式:0—2 min,乙腈由10%提高到30%;2—10 min,乙腈提高至100%;10—13 min,乙腈降至10%,流速为0.5 mL·min−1;进样体积为1 μL;采用电喷雾电离,在正离子模式下采集数据,m/z 扫描范围为50—500 Da。
总铁和Fe2+浓度的测定采用邻菲罗啉分光光度法,Fe3+浓度取两者浓度差;BS浓度测定采用2-硝基苯甲酸显色法;pH计(PHS-3C,上海雷磁)用于溶液pH测定。
-
阿仑尼乌斯定律描述了反应速率常数k与反应温度T的关系,见式(1)。故可根据不同温度条件下,DCF的降解表观反应速率常数,以lnk和1/T作图,并通过对lnk vs 1/T曲线进行拟合进一步计算出体系的表观反应活化能Ea。
式中,k—反应速率常数;A—指前因子;Ea—表观反应活化能(J·mol−1);T—反应的绝对温度(K);R—气体常数(8.314 J·(mol·K)−1)。
-
为评价DCF 在Fe(Ⅲ)/BS体系中的降解效能,本次调查了Fe(Ⅲ)、BS和Fe(Ⅲ)/BS对DCF的降解。结果如图1a所示,单独的Fe(Ⅲ)或BS体系中DCF未被降解,而Fe(Ⅲ)/BS可以降解DCF,其降解规律符合伪一级反应动力学,表观反应速率常数分别为0.7453 min−1。
在Fe(Ⅲ)/BS体系中,Fe(Ⅲ)与BS反应生成金属配体物
{\rm{FeSO}}_3^ + (式(2)),进一步分解为Fe(Ⅱ)和{\rm{SO}}_3^{ \cdot - } (式(3)),其中{\rm{SO}}_3^{ \cdot - } 通过一系列反应生成{\rm{SO}}_4^{ \cdot - } 、{\rm{SO}}_5^{ \cdot - } 和HO•(式(4)—(9)),这些生成的自由基对DCF在Fe(Ⅲ)/BS中的降解起着决定性作用。另外,根据图1b可知,整个反应过程BS在不断的消耗,然而Fe(Ⅲ)在反应时间30s — 3 min浓度基本稳定,这主要是由于Fe(Ⅲ)与BS反应生成的Fe(Ⅱ)可通过式(10)和(11)生成配体物{\rm{FeSO}}_3^ + ,从而实现Fe(Ⅱ/Ⅲ)循环,进而保证体系内自由基持续不断的产生,使DCF在Fe(Ⅲ)/BS体系中得以高效降解。 -
pH值对Fe(Ⅲ)/BS体系的影响可能很大,因为Fe(Ⅲ)、Fe(Ⅱ)和BS在不同pH条件下具有不同的形态,故研究了Fe(Ⅲ)/BS在不同pH值下对DCF的降解,如图2所示。pH 2.0时,由于BS主要以SO2(aq)形式存在,此时该体系不能有效的生成
{\rm{FeSO}}_3^ + 和{\rm{FeHSO}}_3^ + 配体物[36-37],所以DCF几乎没有被降解;pH 4.0时,BS主要以{\rm{HSO}}_3^ - 形式存在,它直接与Fe(Ⅲ)反应生成{\rm{FeSO}}_3^ + 或与Fe(Ⅱ)形成{\rm{FeHSO}}_3^ + 配体物进一步氧化为{\rm{FeSO}}_3^ + ,随后一些活性自由基(如:{\rm{SO}}_3^{ \cdot - } 、{\rm{SO}}_4^{ \cdot - } 、{\rm{SO}}_5^{ \cdot - } 和HO•)在{\rm{FeSO}}_3^ + 分解后生成,最终使DCF得以有效的降解;pH 5.0和pH6.0时DCF几乎未被降解,主要是由于当pH>4.0时,Fe(Ⅲ)会产生沉淀丧失了活化BS的能力。 -
图3描述了不同初始浓度Fe(Ⅲ)对Fe(Ⅲ)/BS体系降解DCF的影响。从降解效率来看,Fe(Ⅲ)浓度为10—80 μmol·L−1时,反应时间内DCF均可有效的降解,且降解效率高于90%;从反应速率来看,当Fe(Ⅲ)浓度低于40 μmol·L−1时,DCF的降解速度随着Fe(Ⅲ)用量的增加而提高,因为随着Fe(Ⅲ)用量增加与BS产生更多的
{\rm{FeSO}}_3^ + ,进而通过一系列反应产生更多的活性自由基。随着Fe(Ⅲ)投加浓度从40 μmol·L−1继续提高至80 μmol·L−1,DCF的降解速度变慢,其原因可解释为体系生成过量的Fe(Ⅱ)会通过式(12)和式(13)捕获{\rm{SO}}_4^{ \cdot - } 和HO•,从而抑制了DCF的降解。 -
图4描述了不同浓度BS对Fe(Ⅲ)/BS体系降解DCF的影响。结果表明,控制Fe(Ⅲ)浓度为10 μmol·L−1不变,当BS浓度由50 μmol·L−1升高至200 μmol·L−1时,DCF的去除率逐渐增加;随着BS浓度提升至400 μmol·L−1时,DCF的降解几乎未变;进一步提升BS的浓度至800 μmol·L−1时,DCF的降解反而受到了抑制。此现象可解释为:BS在一定浓度范围内,随着浓度的提高,一方面体系内直接增加了
{\rm{FeSO}}_3^ + 配体物,另一方面可以加快Fe(Ⅲ)/Fe(Ⅱ)循环,促进溶液中活性自由基的生成;然而过量的BS会通过式(13)和(14)与DCF竞争活性自由基,从而抑制DCF的降解。Xu等[40]在Fe(Ⅲ)/BS体系,Zhang等[24]在Fe(Ⅱ)/BS/irradation体系,Du等[41]在Fe0/BS/O2体系均发现过量的BS可作为自由基捕获剂抑制目标污染物的降解。 -
图5 描述了不同DCF初始浓度在Fe(Ⅲ)、BS 用量分别为10 μmol·L−1 和200 μmol·L−1下,自身浓度的变化对kobs 及降解效率的变化。结果显示,随着DCF初始浓度的升高,其降解效率降低,kobs从1.8223 min−1下降到0.3682 min−1。可见,在相同Fe(Ⅲ)和BS用量一定的条件下,产生的活性物种是一定的,污染物浓度越低越有利于降解。另外,随着DCF的浓度增加,其降解产物的浓度也相应增加,从而使DCF和其降解产物竞争活性自由基。
-
在Fe(Ⅲ)/BS体系中,溶解氧可通过式(3)和(10)影响
{\rm{SO}}_5^{ \cdot - } 和{\rm{FeSO}}_3^ + 的形成,因此可能会影响污染物的去除。为了研究溶解氧对Fe(Ⅲ)/BS体系中DCF降解的影响,分别采用了鼓入空气或氮气对反应溶液进行曝气和保持自然搅拌等3种条件,结果如图6所示。DCF在鼓入空气溶液中的降解与自然搅拌条件下的降解曲线几乎相同,表明溶解氧的浓度不作为DCF降解的限制条件,Huss等[44]在研究Fe(Ⅱ)与BS的反应动力学中也发现此现象。鼓入氮气条件下,虽然DCF的降解受到抑制,但仍有较好的降解趋势,可能是由于1)具有还原性的{\rm{SO}}_3^{ \cdot - } (其标准还原电位−0.73 V)攻击DCF的C−Cl基团发生脱氯反应[45];2){\rm{SO}}_3^{ \cdot - } 具有一定氧化性可攻击有机物的亲电反应位点与之发生氧化反应(反应二级速率常数约为106—107 L·mol−1·s−1)。Zhou等[28]也发现不论是有氧还是厌氧条件下,AO7均可在Fe(Ⅲ)/BS下发生降解,且AO7与{\rm{SO}}_3^{ \cdot - } 的反应二级速率常数为107 L·mol−1·s−1。 -
为了考察温度对Fe(Ⅲ)/BS体系的影响及该体系降解DCF的表观反应活化能,选取15—35 ℃条件下进行,如图7所示。可以看出,随着反应温度升高,DCF的降解速率加快,反应温度15 ℃时,其一级反应速率常数kobs为0.1328 min−1;当反应温度提升至35 ℃时,其kobs增加至2.6078 min−1。可以看出,温度的升高可加快
{\rm{FeSO}}_3^ + 的形成和分解,使体系产生更多的活性自由基;同时温度升高可加快分子的运动速度,使单位时间内分子间的有效碰撞次数增加,对氧化DCF有利。根据阿累尼乌斯(Arrhenius)方程计算了DCF在Fe(Ⅲ)/BS体系中的反应活化能为104.80 kJ·mol−1。 -
Cl− 是水中常见的阴离子,在
{\rm{SO}}_4^{ \cdot - } 氧化降解有机物中起着重要的作用,可通过与{\rm{SO}}_4^{ \cdot - } 反应生成各种含氯自由基,从而影响污染物的降解效率。目前Cl−对BS降解有机物的影响鲜有报道,故本次考察了其对DCF 降解的影响,如图8a 所示。Cl−对DCF降解有一定的抑制作用,且随Cl−浓度增加抑制效果增大,这可能是由于在Fe(Ⅲ)/BS体系中,Cl−可淬灭{\rm{SO}}_4^{ \cdot - } 生成Cl•和{\rm{Cl}}_2^{ \cdot - } ,而BS属还原剂,Cl•又可以氧化体系中的BS,从而减少了{\rm{FeSO}}_3^ + 的产量也一定程度上消耗了活性自由基。图8b描述了不同浓度FA对DCF在Fe(Ⅲ)/BS体系中降解的影响。结果可知,0.5 mg·L−1的FA对DCF的降解有轻微的抑制作用,随着FA浓度的增加,对DCF降解的抑制作用越明显;当FA浓度增加至10 mg·L−1时,DCF的降解完全被抑制。可能有两种原因:1)FA与DCF竞争活性自由基,随着浓度越高竞争越明显;2)FA与铁离子发生络合,减少了溶液中铁离子的含量,同样随着浓度越高对铁离子的络合作用越明显。
-
在Fe(Ⅲ)/BS体系中,可以生成多种活性自由基,如
{\rm{SO}}_3^{ \cdot - } 、{\rm{SO}}_4^{ \cdot - } 、{\rm{SO}}_5^{ \cdot - } 和HO•[30,40]。为明确体系中DCF降解的主要反应自由基,添加了甲醇(MeOH)、叔丁醇(TBA)两种自由基清除剂进行实验。TBA与HO•的二级反应速率常数(7.6 × 108 L·mol−1·s−1)与{\rm{SO}}_4^{ \cdot - } 的(4.0 × 105 L·mol−1·s−1)[46]差3个数量级,故常用作HO•的淬灭剂;而MeOH与{\rm{SO}}_4^{ \cdot - } 和HO•的二级反应速率常数高且相近,分别为2.5 × 107、9.7 × 108 L·mol−1·s−1 [47],同时用作{\rm{SO}}_4^{ \cdot - } 和HO•的淬灭剂;TBA和MeOH的加入还可以区分{\rm{SO}}_3^{ \cdot - } /{\rm{SO}}_5^{ \cdot - } 和{\rm{SO}}_4^{ \cdot - } /HO•,因为两者与{\rm{SO}}_3^{ \cdot - } /{\rm{SO}}_5^{ \cdot - } 的二级反应速率非常低(< 103 L·mol−1·s−1)[46]。为了进一步自由基清除剂与DCF对自由基的竞争反应,引入了ck值[48](c为体系中反应物的摩尔浓度,k为体系中具有活性自由基的反应种类的二级速率常数),见表1。结合表1和图9可知,10 mmol·L−1的TBA与HO•的ck值远大于DCF的,然而10 mmol·L−1的TBA与
{\rm{SO}}_4^{ \cdot - } 的ck值远小于DCF的(ck TBA, HO· = 7.6 × 106 s−1>> ck DCF, HO• = 7.5 × 104 s−1;c{k_{{\rm{TBA,SO}}_4^{ \cdot - }}} = 4.0× 103 s−1<<c{k_{{\rm{DCF,SO}}_4^{ \cdot - }}} = 9.2 × 104 s−1)。这表明在反应体系中加入10 mmol·L−1 TBA只能完全终止HO•,而几乎不能淬灭{\rm{SO}}_4^{ \cdot - } 。此时,DCF的降解的几乎不受影响,表明HO•不是该体系的主要活性自由基。当TBA浓度超过100 mmol·L−1时,DCF的抑制作用明显,此时TBA与{\rm{SO}}_4^{ \cdot - } 的ck值接近甚至超过DCF,故形成竞争反应。同理,对比100 mmol·L−1 TBA和MeOH条件下(c{k_{{\rm{TBA,SO}}_4^{ \cdot - }}} = 4.0× 104 s−1 <<c{k_{{\rm{MeOH,SO}}_4^{ \cdot - }}} ),DCF去除率的差值可归因于{\rm{SO}}_4^{ \cdot - } 的氧化作用。体系中加入500 mmol·L−1 Me时,可完全淬灭{\rm{SO}}_4^{ \cdot - } 和 HO•,因为500 mmol·L−1 Me和{\rm{SO}}_4^{ \cdot - } 或 HO•的ck值均远大于DCF的;然而,仍有11%的DCF被去除,表明体系中还可能存在{\rm{SO}}_5^{ \cdot - } ,但其不是主要活性自由基。综上,{\rm{SO}}_4^{ \cdot - } 和{\rm{SO}}_5^{ \cdot - } 都可能是Fe(Ⅲ)/BS降解DCF的原因,而{\rm{SO}}_4^{ \cdot - } 是主要的反应物种。 -
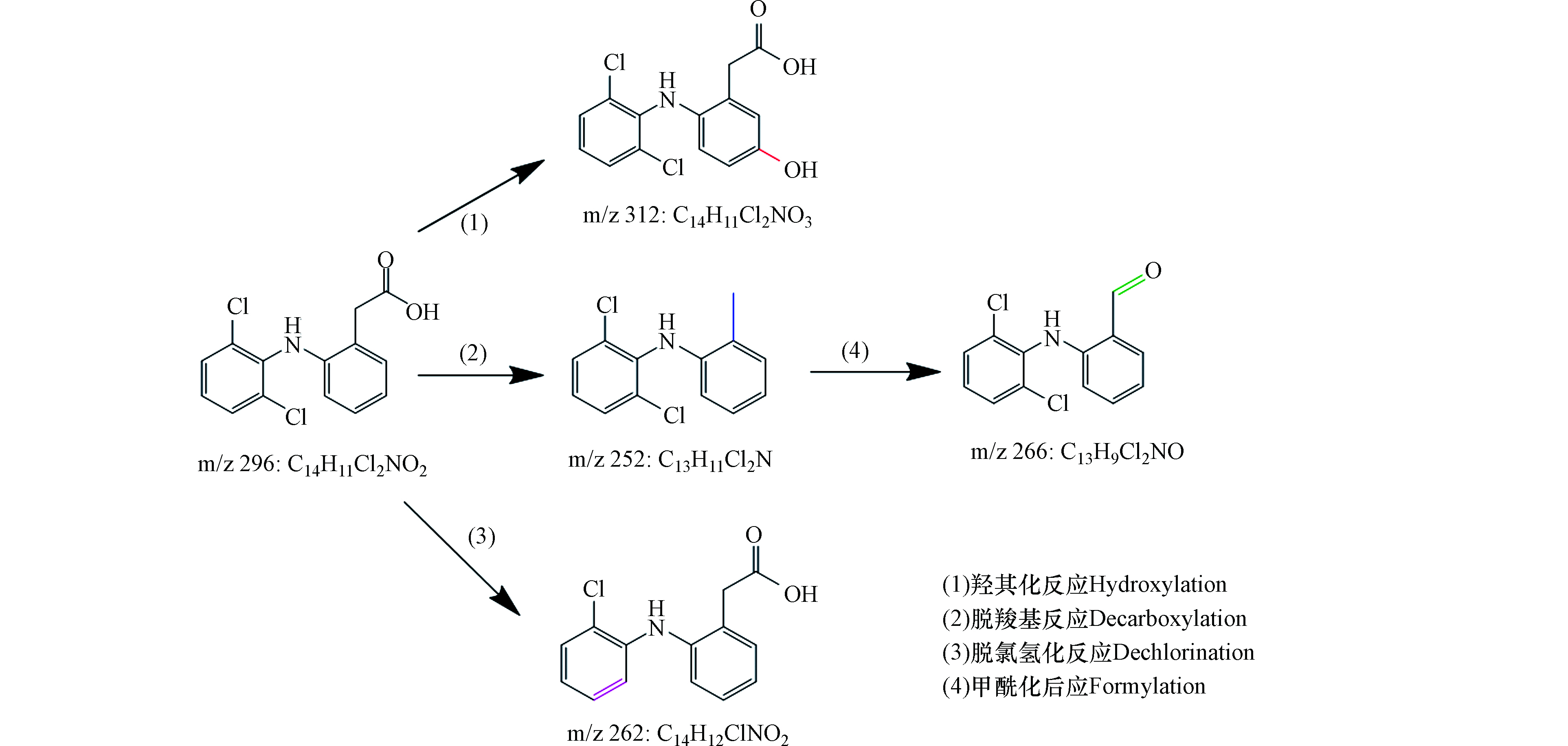
在Fe(Ⅲ)/BS降解DCF的过程中,共检测到4种转化产物,如表2所示。基于此提出了DCF在该体系中可能的反应机理,主要包括:(1)羟基化反应、(2)脱羧基反应、(3)脱氯氢化反应、(4)甲酰化反应,如图10所示。DCF可通过途径(1)、(2)和(3)分别生成产物m/z 312、252和262,产物m/z 252 通过途径(4)进一步转化为产物m/z 266。
-
(1)Fe(Ⅲ)/BS可实现Fe(Ⅱ/Ⅲ)自循环,快速高效地降解DCF,其降解规律符合伪一级反应动力学,表观速率常数为0.7453 s−1;表观反应活化能为104.80 kJ·mol−1。
(2)初始pH为4.0时,DCF的降解效率最佳;其降解随着Fe(Ⅲ)和BS的用量增加而增加,然而过量的Fe(Ⅲ)或BS可抑制DCF降解。
(3)DCF在Fe(Ⅲ)/BS的降解与溶解氧浓度无关,在自然搅拌、曝空气或氮气的条件下均可有效的降解;Cl−和FA对DCF的降解起到抑制作用,且两者浓度越高,抑制作用越明显。
(4)Fe(Ⅲ)/BS体系中可能存在
{\rm{SO}}_4^{ \cdot - } 和{\rm{SO}}_5^{ \cdot - } 两种活性自由基,而{\rm{SO}}_4^{ \cdot - } 是主要的反应物种。DCF可能的降解机理包括:羟基化、脱羧基、脱氯氢化和甲酰化反应等4种不同的反应路径。
Fe(Ⅲ)/HSO3−降解双氯芬酸钠动力学及机理研究
Study on kinetics and mechanism of diclofenac degradation by ferric activated Bisulfite
-
摘要: 研究了基于Fe(Ⅲ)活化亚硫酸氢盐(
HSO−3 ,BS)的新型高级氧化技术对水中双氯芬酸钠的降解,考察了pH、Fe3+用量、BS用量、DCF初始浓度、溶解氧、温度、Cl−及黄腐酸(FA)对Fe(Ⅲ)/BS降解DCF的影响,探讨了DCF的降解机理。结果表明, Fe(Ⅲ)/BS可实现Fe(Ⅲ/Ⅱ)自循环,快速高效地降解DCF,其降解符合伪一级反应动力学,表观速率常数为0.7453 s−1,表观反应活化能为104.80 kJ·mol−1;该体系SO⋅−4 起主要作用。初始pH 4.0时,DCF的降解效率最佳;其降解随Fe(Ⅲ)和BS用量增加而增加,然而过量的Fe(Ⅲ)或BS则呈现抑制作用;在自然搅拌、曝空气或氮气条件下DCF均可得到有效降解;Cl−和黄腐酸(FA)对DCF的降解有抑制作用,其抑制效果随两者浓度增加而增大。产物鉴定表明,DCF可能的4种降解路径为:羟基化、脱羧基、脱氯氢化和甲酰化反应。Abstract: In this study, a new advanced oxidation technology based on Fe(Ⅲ) activated bisulfite (BS) was employed to degrade diclofenac in water. The effects of pH, Fe3+ dosage, BS dosage, dissolved oxygen, temperature and fulvic acid (FA) on DCF degradation by Fe(Ⅲ)/BS were investigated. The degradation mechanism of DCF was also discussed. The results showed that Fe(Ⅲ)/BS realized the degradation of DCF rapidly and efficiently, along with the self-circulation of Fe(Ⅲ/Ⅱ). The DCF degradation conformed to the pseudo-first-order reaction kinetics, with the apparent rate constant of 0.7453 s−1 and the apparent reaction activation energy of 104.80 kJ·mol−1. In this system,SO⋅−4 played the dominated role. The optimal degradation pH was 4.0, and the degradation of DCF increased with the increase of the amount of Fe(Ⅲ) and BS, while excessive Fe(Ⅲ) or BS would inhibit thus as aSO⋅−4 scavenger. DCF could be effectively degraded under stirring, aeration or nitrogen by Fe(Ⅲ)/BS, showing that the degradation reaction was zero order with respect to oxygen. FA and Cl− could inhibit the degradation of DCF, and the inhibitory effect was proportional to the concentration of FA or Cl−. Based on these identified by-products, the probable degradation mechanism of DCF was proposed, including hydroxylation, decarboxylation, dichlorination-hydrogenation and formylation.-
Key words:
- diclofenac /
- bisulfite /
- ferric /
- sulfate radical /
- degradation
-
药物和个人护理产品(PPCPs)是一类 “新兴”环境污染物,它们具有在低剂量下诱导生物体产生生理效应的能力,作为医用或保健药品被广泛应用于人或动物[1]。然而,这些药物被人体或动物摄入后,仅少部分发生代谢,约30%—90%以原形药物排出体外,并不断释放到水环境中[2]。作为典型的PPCPs,双氯芬酸钠(diclofenac,DCF)是全球目前最畅销的解热镇痛消炎药物之一,每天约被世界各地3000万人广泛使用,目前已在各水环境中检出,如:污水处理厂、地表水、地下水、甚至自来水中,浓度在ng·L−1 — μg·L−1之间[3-4]。研究表明,痕量的DCF便可引起水生生物内脏器官发生病变[5-6],并通过食物链积累对生态系统和人类健康构成潜在威胁[7],其带来的环境污染问题也受到国内外广泛关注[8-10]。与大多数PPCPs相同,DCF难以被生物降解[11-12];目前,国内外主要采用化学氧化法(Fenton氧化法[13]、光化学氧化法[14-16]、电化学氧化法[17]、臭氧氧化法[18]、超声波辐射法[19-20]和光子束流辐射[21])对其降解。
近年来基于亚硫酸盐(S(Ⅳ))产生硫酸根自由基的高级氧化技术受到环境工作者关注,但主要用于大气治理和烟气脱硫,在水处理领域的应用研究仍处于起步阶段。从经济和毒性角度上看,亚硫酸盐较过硫酸盐具有价格低廉、半致死浓度更高等优点。目前,活化S(Ⅳ) 产生硫氧根自由基的方法主要有:UV辐射活化[22-23]和过渡金属活化[24-26]等。鉴于,Fe(Ⅲ)自然界中含量广泛,价格便宜、环境友好等特点,Fe(Ⅲ)活化S(Ⅳ)降解有机物具有应用及推广潜力。Yuan等[27]采用Fe(Ⅲ)/S(Ⅳ)降解苯胺,研究了不同浓度下反应剂对苯胺降解的影响;Zhou等[28]研究了Fe(Ⅲ)/S(Ⅳ)体系中不同硫氧自由基与酸性橙7的二级反应速率常数。现该技术的关注点主要为该体系的最佳反应条件及体系中存在的活性自由基,目标污染物的选取主要为有机染料、苯胺类有机物以及重金属等[29-30]。然而,将Fe(Ⅲ)/S(Ⅳ)作为高级氧化体系降解PPCPs类新兴有机物的研究报道较少。
本研究选用Fe(Ⅲ)活化亚硫酸氢钠(BS)处理DCF模拟废水,除了分析反应试剂对其降解的影响,还考察了水中常见干扰离子氯离子及黄腐酸(FA)的影响;并基于温度的影响计算了Fe(Ⅲ)/S(Ⅳ)体系的表观反应活化能;通过自由基抑制实验分析体系中的活性自由基;最后利用LC_MC测定DCF的降解中间产物,推测其可能的降解机理。
1. 实验部分(Experimental section)
1.1 试剂
双氯芬酸钠(C14H10Cl2NO2Na,纯度≥98%)、1,10-菲罗啉(分析纯)、2-硝基苯甲酸和黄腐酸(FA,纯度≥90%)购于Aladdin公司;亚硫酸氢钠(NaHSO3)、九水硝酸铁(Fe(NO3)3·9H2O)、磷酸二氢钾(KH2PO4)、磷酸氢二钠(Na2HPO4)、氢氧化钠(NaOH)、硫代硫酸钠(Na2S2O3)、氯化钠(NaCl)、硫酸(H2SO4)和无水乙酸钠(CH3COONa)为分析纯,冰乙酸和叔丁醇为色谱纯,上述药品购于成都科龙化学品有限公司;甲醇(色谱纯)购于Fisher Scientific公司。去离子水(18MΩ·cm)用于试剂的溶解和配置。
1.2 实验方法
实验在250 mL烧杯中进行,通过水浴恒温磁力搅拌装置来控制搅拌速度和反应温度,其中转子转速为600 r·min−1,温度设置为25 ℃(温度影响除外)。配置100 mL一定浓度的DCF溶液,投加一定量Fe(NO3)3储备溶液,然后用0.2 mol·L−1 NaOH和0.2 mol·L−1 H2SO4溶液将其pH值调节至预设值附近,投加1 mL NaHSO3储备溶液启动反应,反应过程中不对溶液pH进行控制。分别在0、0.5、1、2、3、5 min取1 mL样品放入装有50 μL的0.4 mol硫代硫酸钠的液相小瓶,快速摇匀终止反应后待测。所有实验至少进行两次,取平均值计算去除率。为确保加入NaHSO3后反应初始pH在设定值± 0.05范围内,需在投加Fe(NO3)3溶液后对pH进行多次微调和投加NaHSO3溶液预实验来确定预设值[31],除pH影响实验外,初始反应pH值均为4.0。除产物检测,所有实验至少进行2次,取平均值计算去除率。
1.3 分析方法
DCF浓度测定采用高效液相色谱仪(Waters 2695)。具体参数:固定相为C18柱(5 μm × 4.6 mm × 150 mm);流动相由1‰乙酸水溶液与甲醇(25:75,V/V)组成,流速为1 mL·min−1;检测波长276 nm;进样体积为20 μL;柱温为30 ℃。
DCF中间产物的检测采用UPLC-QTOT-MS(Waters)。具体参数:色谱柱为BEH C18(1.7 μm × 2.1 mm × 50 mm);流动相为乙腈和水(0.1%甲酸),采用梯度洗脱的方式:0—2 min,乙腈由10%提高到30%;2—10 min,乙腈提高至100%;10—13 min,乙腈降至10%,流速为0.5 mL·min−1;进样体积为1 μL;采用电喷雾电离,在正离子模式下采集数据,m/z 扫描范围为50—500 Da。
总铁和Fe2+浓度的测定采用邻菲罗啉分光光度法,Fe3+浓度取两者浓度差;BS浓度测定采用2-硝基苯甲酸显色法;pH计(PHS-3C,上海雷磁)用于溶液pH测定。
1.4 表观活化能计算
阿仑尼乌斯定律描述了反应速率常数k与反应温度T的关系,见式(1)。故可根据不同温度条件下,DCF的降解表观反应速率常数,以lnk和1/T作图,并通过对lnk vs 1/T曲线进行拟合进一步计算出体系的表观反应活化能Ea。
lnk=lnA−Ea/RT (1) 式中,k—反应速率常数;A—指前因子;Ea—表观反应活化能(J·mol−1);T—反应的绝对温度(K);R—气体常数(8.314 J·(mol·K)−1)。
2. 结果与讨论(Results and discussion)
2.1 Fe(Ⅲ)/BS 降解DCF
为评价DCF 在Fe(Ⅲ)/BS体系中的降解效能,本次调查了Fe(Ⅲ)、BS和Fe(Ⅲ)/BS对DCF的降解。结果如图1a所示,单独的Fe(Ⅲ)或BS体系中DCF未被降解,而Fe(Ⅲ)/BS可以降解DCF,其降解规律符合伪一级反应动力学,表观反应速率常数分别为0.7453 min−1。
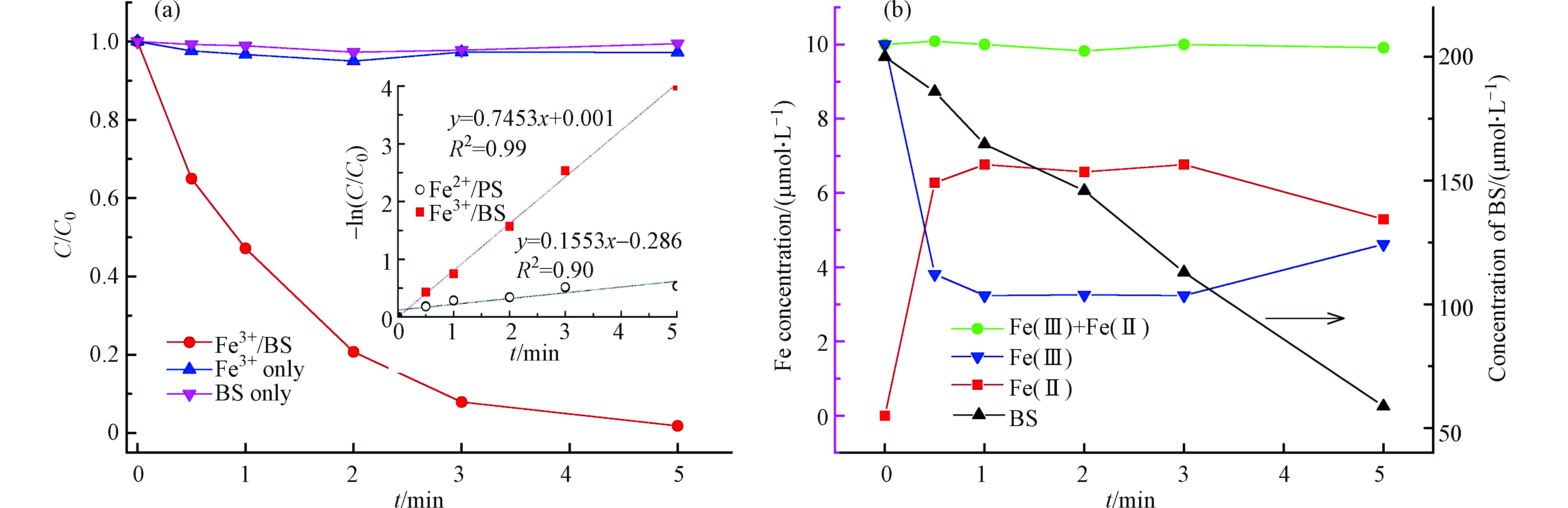 图 1 DCF在不同体系的降解(a),Fe(Ⅲ)/BS中Fe和BS浓度变化(b)Figure 1. Degradation of DCF in different systems (a) and the variation of Fe(Ⅱ), Fe(Ⅲ) and S(Ⅳ) concentrations in Fe(III)/BS system (b).pH = 4.0, [DCF] = 10 μmol·L−1, [Fe3+] =10 μmol·L−1, [BS] = 0.2 mmol·L−1, [PS] = 1 mmol·L−1.
图 1 DCF在不同体系的降解(a),Fe(Ⅲ)/BS中Fe和BS浓度变化(b)Figure 1. Degradation of DCF in different systems (a) and the variation of Fe(Ⅱ), Fe(Ⅲ) and S(Ⅳ) concentrations in Fe(III)/BS system (b).pH = 4.0, [DCF] = 10 μmol·L−1, [Fe3+] =10 μmol·L−1, [BS] = 0.2 mmol·L−1, [PS] = 1 mmol·L−1.在Fe(Ⅲ)/BS体系中,Fe(Ⅲ)与BS反应生成金属配体物
FeSO+3 SO⋅−3 SO⋅−3 SO⋅−4 SO⋅−5 FeSO+3 Fe(Ⅲ)+HSO−3⟶FeSO+3+H+k≈1×103L⋅mol−1⋅s−1[32] (2) FeSO+3⟶Fe(Ⅱ)+SO⋅−3k=0.19s−1[32] (3) {\rm{SO}}_3^{ \cdot - } + {{\rm{O}}_2} \longrightarrow {\rm{SO}}_5^{ \cdot - } \quad \quad k = {\rm{ }}\left( {1.1—2.5} \right){\rm{ }} \times {\rm{ }}{10^9}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[33]} (4) {\rm{SO}}_5^{ \cdot - } + {\rm{ HSO}}_3^ - \longrightarrow {\rm{ SO}}_4^{2 - } + {\rm{ SO}}_4^{ \cdot - } + {\rm{ }}{{\rm{H}}^ + } \quad \quad k = {\rm{ }}1.2{\rm{ }} \times {\rm{ }}{10^4}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[34]} (5) {\rm{SO}}_5^{ \cdot - } + {\rm{ SO}}_3^{ \cdot - } \longrightarrow {\rm{ SO}}_4^{2 - } + {\rm{ SO}}_4^{ \cdot - } \quad \quad k = {\rm{ }}1.2{\rm{ }} \times {\rm{ }}{10^4}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[34]} (6) 2{\rm{SO}}_5^{ \cdot - } \longrightarrow 2{\rm{ SO}}_4^{ \cdot - } + {\rm{ }}2{{\rm{O}}_2} \quad \quad k = {10^4}{\text{—}}{10^8}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[34]} (7) {\rm{SO}}_4^{ \cdot - } + {\rm{ O}}{{\rm{H}}^ - } \longrightarrow {\rm{ SO}}_4^{2 - } + {\rm{ H}}{{\rm{O}}^ \cdot } \quad \quad k = {\rm{ }}\left( {1.4{\text{—}}6.5} \right){\rm{ }} \times {\rm{ }}{10^7}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[35]} (8) {\rm{SO}}_4^{ \cdot - } + {\rm{ }}{{\rm{H}}_{\rm{2}}}{\rm{O }} \longrightarrow {\rm{ SO}}_4^{2 - } + {\rm{ H}}{{\rm{O}}^ \cdot } + {{\rm{H}}^ + } \quad \quad k = {\rm{ }}1.1{\rm{ }} \times {\rm{ }}{10^1}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[35]} (9) {\rm{Fe}}{\left( {{{\text{Ⅱ}}}} \right)^{}} + {\rm{ HSO}}_3^ - \longrightarrow {\rm{ FeHSO}}_3^ + \quad \quad k = {\rm{ }}{10^4}\;{\rm{L\cdot mo}}{{\rm{l}}^{{\rm{ - 1}}}}{\rm{\cdot}}{{\rm{s}}^{{\rm{ - 1}}}}^{[36]} (10) {\rm{FeHSO}}_3^ + + {\rm{ }}1/4{{\rm{O}}_2} \longrightarrow {\rm{ FeSO}}_3^ + + {\rm{ }}1/2{{\rm{H}}_{\rm{2}}}{\rm{O}} \quad \quad k = {\rm{ }}1.69{\rm{ }} \times {10^3}\;{\rm{L\cdot mo}}{{\rm{l}}^{{\rm{ - 1}}}}{\rm{\cdot}}{{\rm{s}}^{{\rm{ - 1}}}}^{[36]} (11) 2.2 Fe(Ⅲ)/BS 体系降解DCF影响因素
2.2.1 pH值的影响
pH值对Fe(Ⅲ)/BS体系的影响可能很大,因为Fe(Ⅲ)、Fe(Ⅱ)和BS在不同pH条件下具有不同的形态,故研究了Fe(Ⅲ)/BS在不同pH值下对DCF的降解,如图2所示。pH 2.0时,由于BS主要以SO2(aq)形式存在,此时该体系不能有效的生成
{\rm{FeSO}}_3^ + {\rm{FeHSO}}_3^ + {\rm{HSO}}_3^ - {\rm{FeSO}}_3^ + {\rm{FeHSO}}_3^ + {\rm{FeSO}}_3^ + {\rm{SO}}_3^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_5^{ \cdot - } {\rm{FeSO}}_3^ + 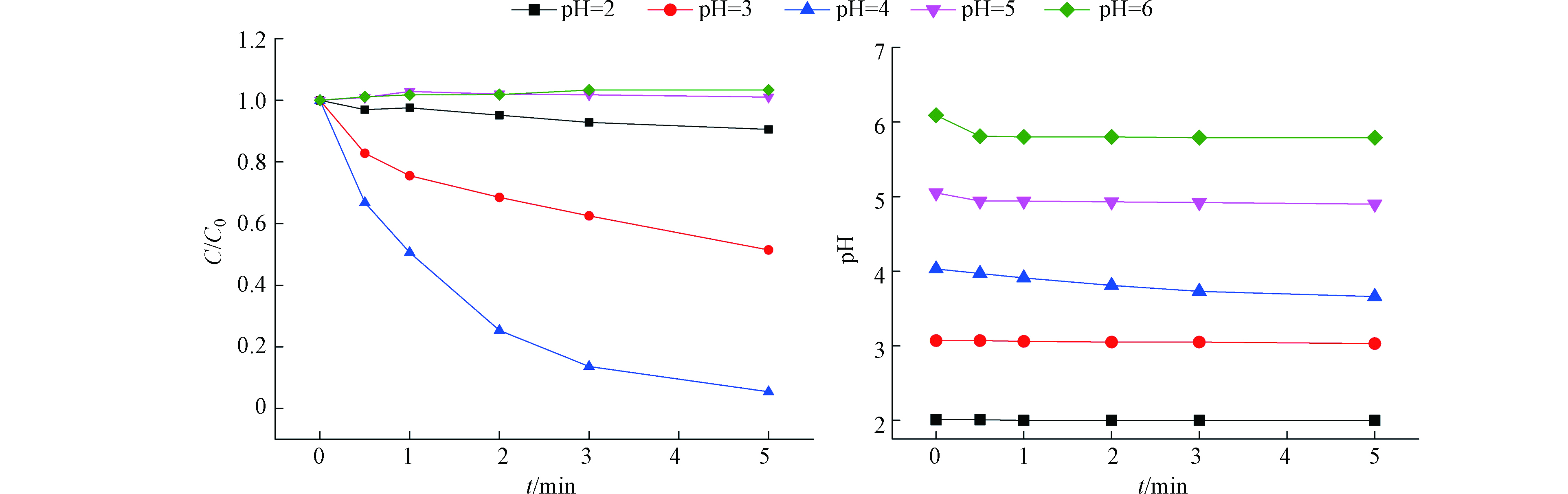 图 2 pH对DCF降解的影响(a),反应过程中pH的变化情况(b)Figure 2. Effect of pH (a) and changes in pH (b) on DCF degradation by Fe(Ⅲ)/BS.[DCF] = 10 μmol·L−1, [Fe3+] = 10 μmol·L−1,[BS] = 0.2 mmol·L−1, T = 25 ℃
图 2 pH对DCF降解的影响(a),反应过程中pH的变化情况(b)Figure 2. Effect of pH (a) and changes in pH (b) on DCF degradation by Fe(Ⅲ)/BS.[DCF] = 10 μmol·L−1, [Fe3+] = 10 μmol·L−1,[BS] = 0.2 mmol·L−1, T = 25 ℃2.2.2 Fe(Ⅲ)浓度的影响
图3描述了不同初始浓度Fe(Ⅲ)对Fe(Ⅲ)/BS体系降解DCF的影响。从降解效率来看,Fe(Ⅲ)浓度为10—80 μmol·L−1时,反应时间内DCF均可有效的降解,且降解效率高于90%;从反应速率来看,当Fe(Ⅲ)浓度低于40 μmol·L−1时,DCF的降解速度随着Fe(Ⅲ)用量的增加而提高,因为随着Fe(Ⅲ)用量增加与BS产生更多的
{\rm{FeSO}}_3^ + {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } + {\rm{ Fe}}\left( {{{\text{Ⅱ}}}} \right){\rm{ }} \longrightarrow {\rm{ Fe}}\left( {{{\text{Ⅲ}}}} \right){\rm{ }} + {\rm{ SO}}_4^{2 - } \quad \quad k = {\rm{ }}4.6{\rm{ }} \times {\rm{ }}{10^9}\; {\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[38]} (12) {\rm{H}}{{\rm{O}}^ \cdot } + {\rm{ Fe}}\left( {{{\text{Ⅱ}}}} \right){\rm{ }} \longrightarrow {\rm{ Fe}}\left( {{{\text{Ⅲ}}}} \right){\rm{ }} + {\rm{ O}}{{\rm{H}}^ - } \quad \quad k = {\rm{ }}4.3{\rm{ }} \times {\rm{ }}{10^8}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[39]} (13) 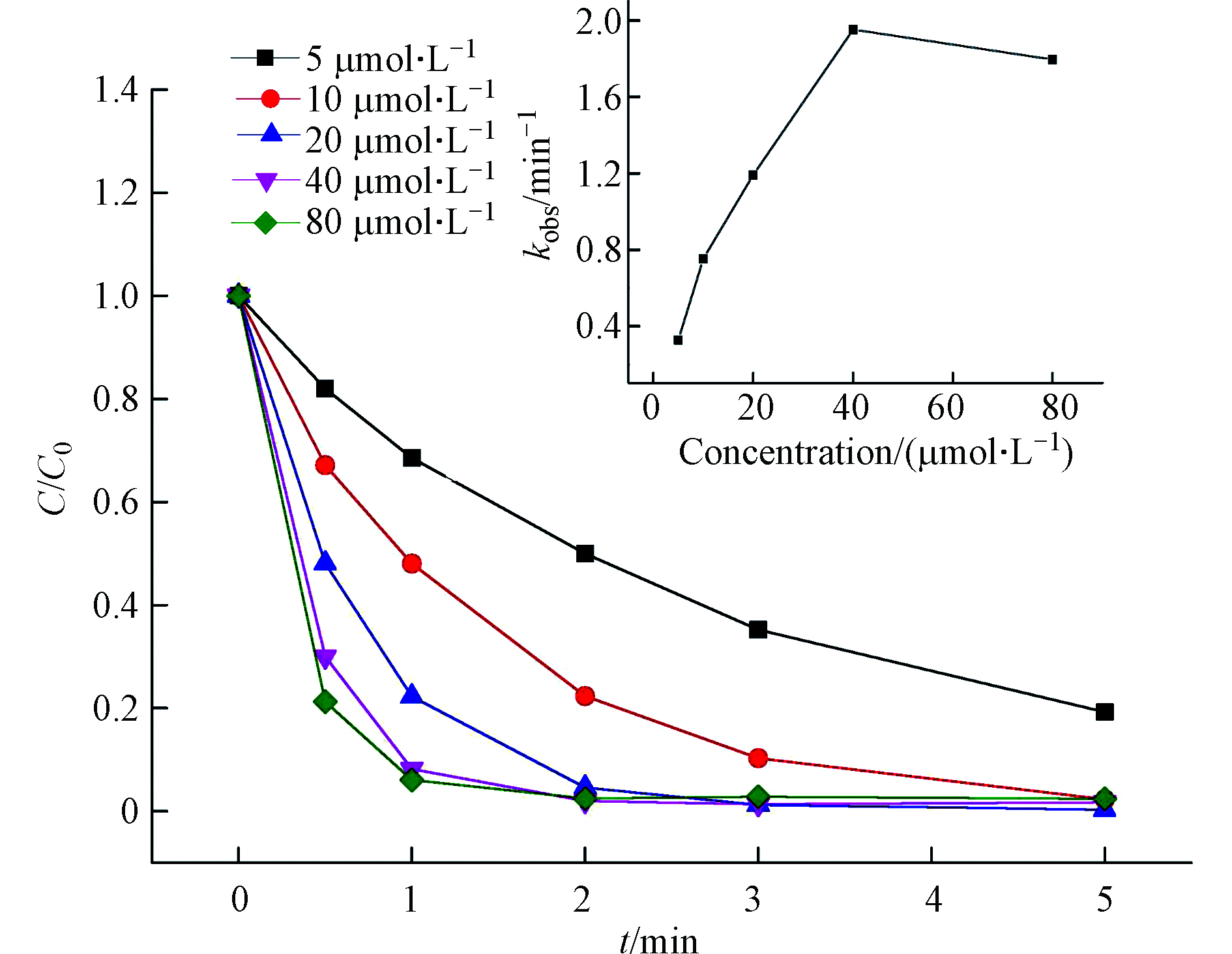 图 3 Fe(Ⅲ)浓度对DCF降解的影响Figure 3. Effect of Fe(Ⅲ) dosage on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = 10 μmol·L−1, [BS] = 0.2 mmol·L−1, T = 25 ℃, reaction time 5 min
图 3 Fe(Ⅲ)浓度对DCF降解的影响Figure 3. Effect of Fe(Ⅲ) dosage on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = 10 μmol·L−1, [BS] = 0.2 mmol·L−1, T = 25 ℃, reaction time 5 min2.2.3 BS浓度的影响
图4描述了不同浓度BS对Fe(Ⅲ)/BS体系降解DCF的影响。结果表明,控制Fe(Ⅲ)浓度为10 μmol·L−1不变,当BS浓度由50 μmol·L−1升高至200 μmol·L−1时,DCF的去除率逐渐增加;随着BS浓度提升至400 μmol·L−1时,DCF的降解几乎未变;进一步提升BS的浓度至800 μmol·L−1时,DCF的降解反而受到了抑制。此现象可解释为:BS在一定浓度范围内,随着浓度的提高,一方面体系内直接增加了
{\rm{FeSO}}_3^ + {\rm{SO}}_4^{ \cdot - } + {\rm{ HSO}}_3^ - \longrightarrow {\rm{ SO}}_3^{ \cdot - } + {\rm{ SO}}_4^{2 - } + {{\rm{H}}^ + }\quad \quad k = {\rm{ }}1.3 \times {10^8}-{\rm{ }}2.5{\rm{ }} \times {\rm{ }}{10^9}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[42 - 43]} (14) {\rm{H}}{{\rm{O}}^ \cdot } + {\rm{ HSO}}_3^ - \longrightarrow {\rm{ SO}}_3^{ \cdot - } + {\rm{ }}{{\rm{H}}_{\rm{2}}}{\rm{O}}\quad \quad k = {\rm{ }}4.5{\rm{ }} \times {\rm{ }}{10^9}\;{\rm{L\cdot mo}}{{\rm{l}}^{ - 1}}\cdot{{\rm{s}}^{ - 1}}^{[24]} (15)  图 4 BS浓度对DCF降解的影响Figure 4. Effect of BS dosage on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, T = 25 ℃
图 4 BS浓度对DCF降解的影响Figure 4. Effect of BS dosage on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, T = 25 ℃2.2.4 DCF初始浓度的影响
图5 描述了不同DCF初始浓度在Fe(Ⅲ)、BS 用量分别为10 μmol·L−1 和200 μmol·L−1下,自身浓度的变化对kobs 及降解效率的变化。结果显示,随着DCF初始浓度的升高,其降解效率降低,kobs从1.8223 min−1下降到0.3682 min−1。可见,在相同Fe(Ⅲ)和BS用量一定的条件下,产生的活性物种是一定的,污染物浓度越低越有利于降解。另外,随着DCF的浓度增加,其降解产物的浓度也相应增加,从而使DCF和其降解产物竞争活性自由基。
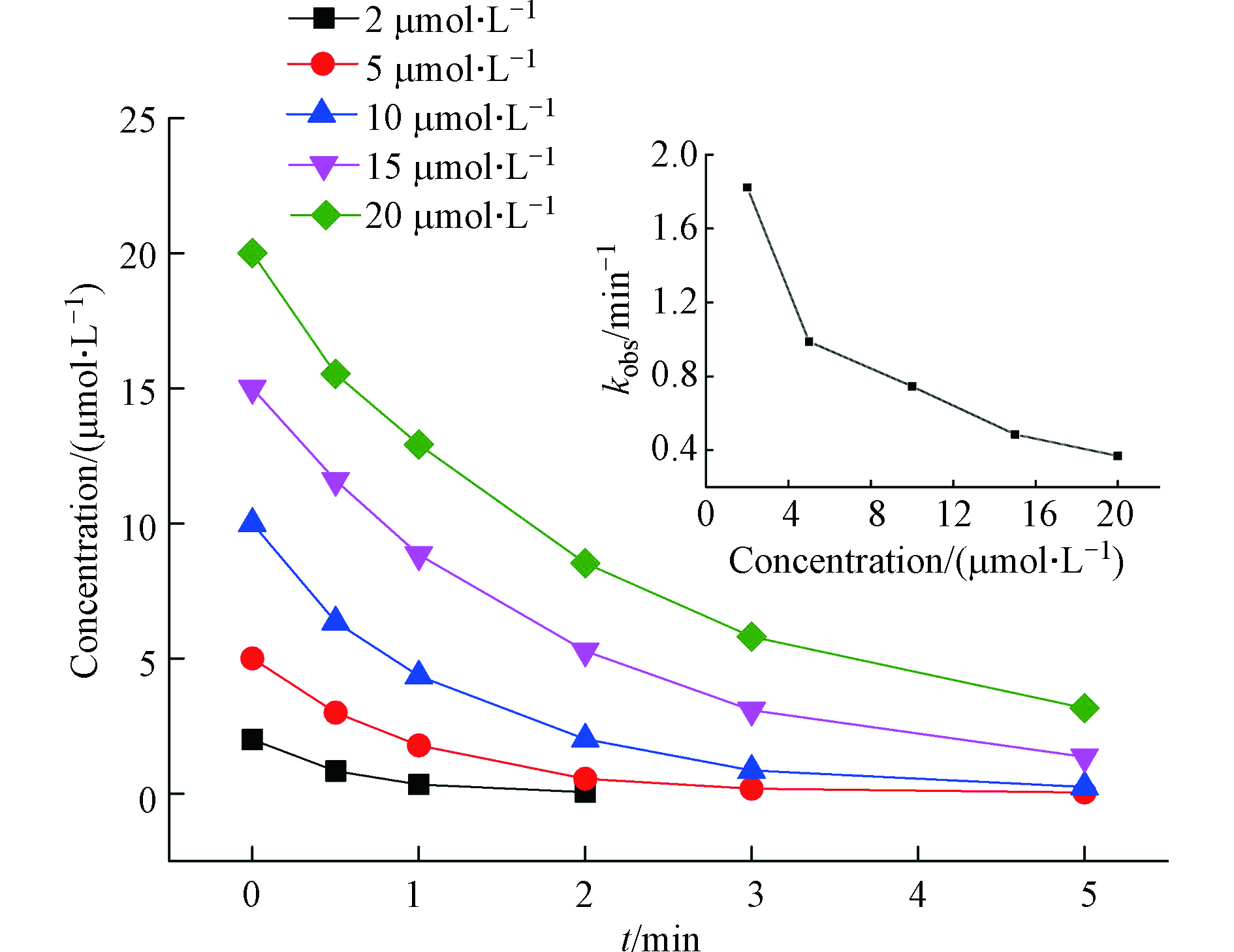 图 5 DCF初始浓度对其自身降解的影响Figure 5. Effect of initial concentration of DCF on its own degradation in Fe(Ⅲ)/BS system.pH =4.0, [Fe3+] = 10 μmol·L−1, BS = 200 μmol·L−1, T = 25 ℃
图 5 DCF初始浓度对其自身降解的影响Figure 5. Effect of initial concentration of DCF on its own degradation in Fe(Ⅲ)/BS system.pH =4.0, [Fe3+] = 10 μmol·L−1, BS = 200 μmol·L−1, T = 25 ℃2.2.5 溶解氧的影响
在Fe(Ⅲ)/BS体系中,溶解氧可通过式(3)和(10)影响
{\rm{SO}}_5^{ \cdot - } {\rm{FeSO}}_3^ + {\rm{SO}}_3^{ \cdot - } {\rm{SO}}_3^{ \cdot - } {\rm{SO}}_3^{ \cdot - }  图 6 溶解氧的影响Figure 6. Effect of dissolved oxygen on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, T = 25 ℃
图 6 溶解氧的影响Figure 6. Effect of dissolved oxygen on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, T = 25 ℃2.2.6 温度的影响
为了考察温度对Fe(Ⅲ)/BS体系的影响及该体系降解DCF的表观反应活化能,选取15—35 ℃条件下进行,如图7所示。可以看出,随着反应温度升高,DCF的降解速率加快,反应温度15 ℃时,其一级反应速率常数kobs为0.1328 min−1;当反应温度提升至35 ℃时,其kobs增加至2.6078 min−1。可以看出,温度的升高可加快
{\rm{FeSO}}_3^ + 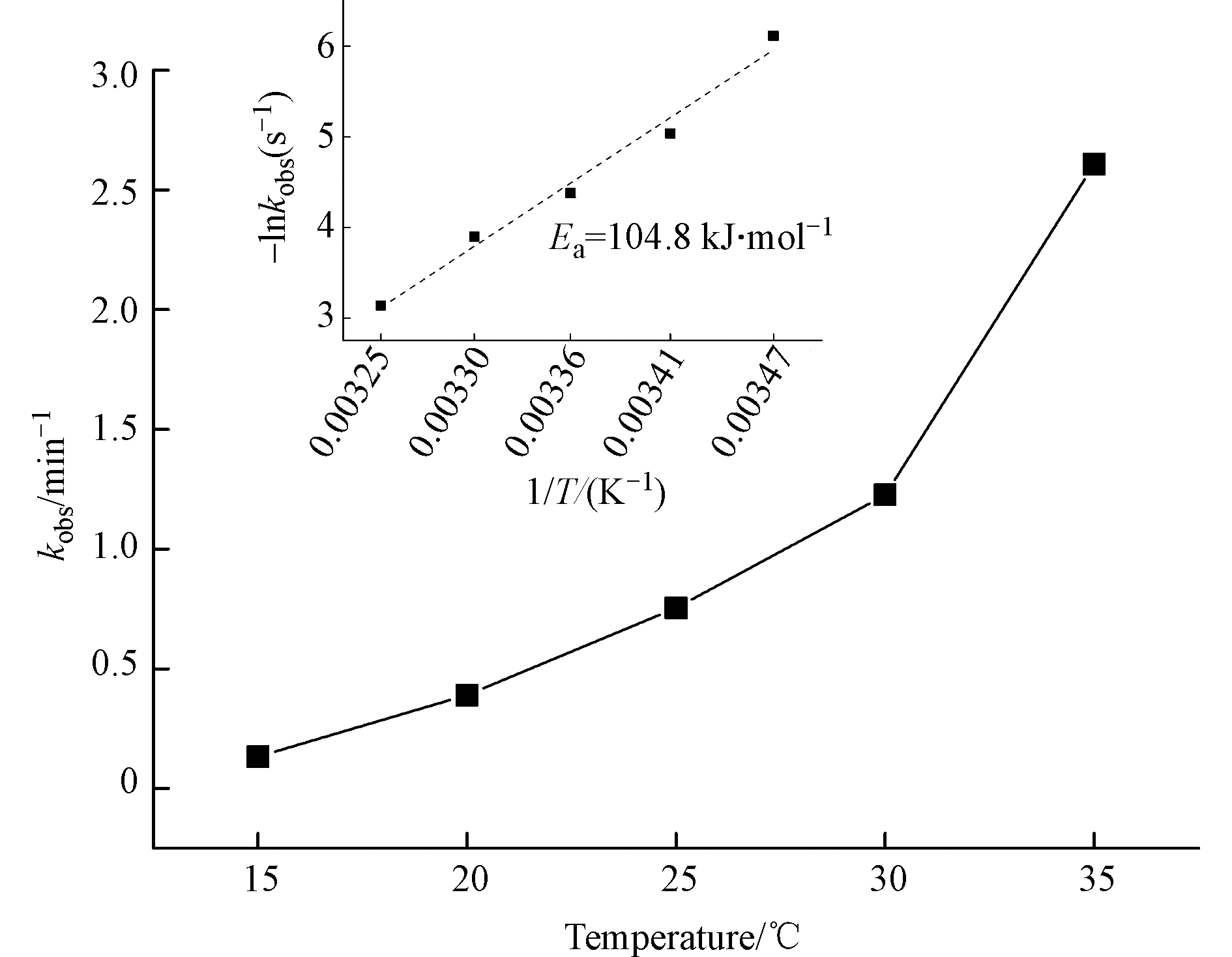 图 7 温度对DCF降解的影响Figure 7. Effect of reaction temperature on kobs in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, [BS] = 0.2 mmol·L−1
图 7 温度对DCF降解的影响Figure 7. Effect of reaction temperature on kobs in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, [BS] = 0.2 mmol·L−12.3 Cl− 和FA的影响
Cl− 是水中常见的阴离子,在
{\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{Cl}}_2^{ \cdot - } {\rm{FeSO}}_3^ + 图8b描述了不同浓度FA对DCF在Fe(Ⅲ)/BS体系中降解的影响。结果可知,0.5 mg·L−1的FA对DCF的降解有轻微的抑制作用,随着FA浓度的增加,对DCF降解的抑制作用越明显;当FA浓度增加至10 mg·L−1时,DCF的降解完全被抑制。可能有两种原因:1)FA与DCF竞争活性自由基,随着浓度越高竞争越明显;2)FA与铁离子发生络合,减少了溶液中铁离子的含量,同样随着浓度越高对铁离子的络合作用越明显。
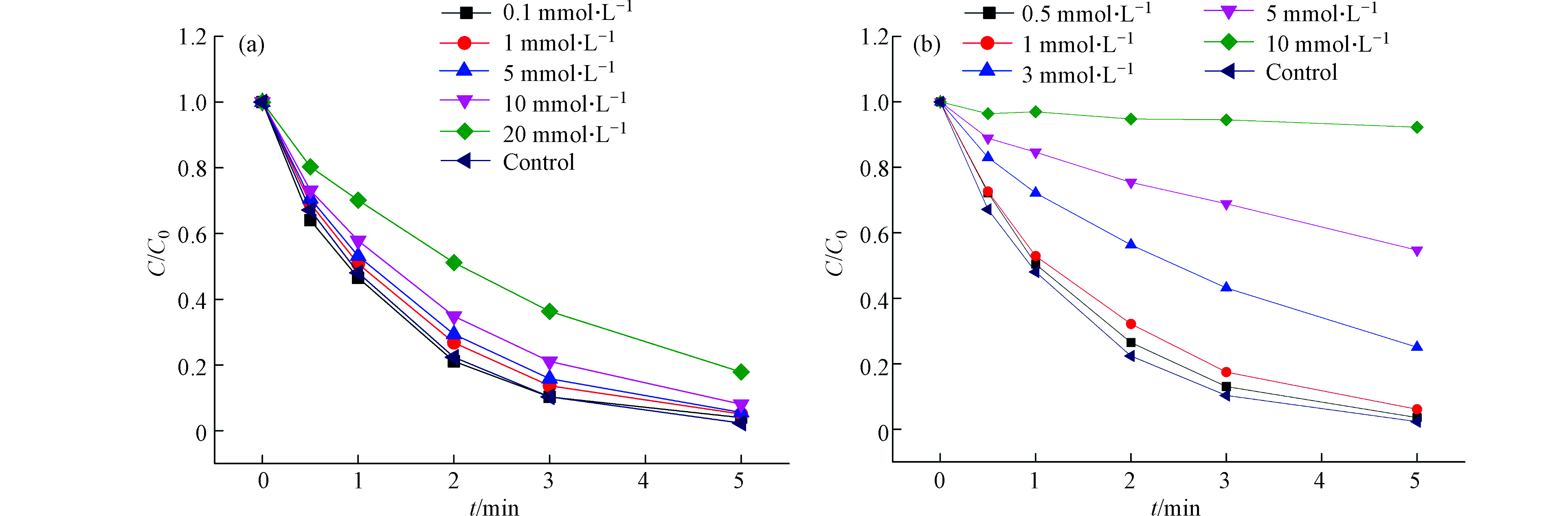 图 8 氯离子(a)和黄腐酸(b)对DCF降解的影响Figure 8. Effect of Cl− (a) and FA (b) on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, [BS] = 0.2 mmol·L−1, T = 25 ℃
图 8 氯离子(a)和黄腐酸(b)对DCF降解的影响Figure 8. Effect of Cl− (a) and FA (b) on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, [BS] = 0.2 mmol·L−1, T = 25 ℃2.4 自由基抑制实验
在Fe(Ⅲ)/BS体系中,可以生成多种活性自由基,如
{\rm{SO}}_3^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_5^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_3^{ \cdot - } {\rm{SO}}_5^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_3^{ \cdot - } {\rm{SO}}_5^{ \cdot - } 表 1 目标物与反应自由基的ck值Table 1. The ck values of target and reactive radical目标物Reactive species 浓度/(mol·L−1)Concentration HO• {\rm{SO}}_4^{ \cdot - } c/ (L·mol−1·s−1) ck /s−1 k /(L·mol−1·s−1) ck /s−1 DCF 1.0 × 10−5 7.5 × 109 7.5 × 104 9.2 × 109 9.2 × 104 TBA 1.0 × 10−2 7.6 × 108 7.6 × 106 4.0 × 105 4.0 × 103 1.0 × 10−1 7.6 × 107 4.0 × 104 0.5 3.8 × 108 2.0 × 105 MeOH 1.0 × 10−1 9.7 × 108 9.7 × 107 2.5 × 107 2.5 × 106 0.5 4.8 × 108 1.3 × 107 | Show Table DownLoad:
CSV
DownLoad:
CSV
结合表1和图9可知,10 mmol·L−1的TBA与HO•的ck值远大于DCF的,然而10 mmol·L−1的TBA与
{\rm{SO}}_4^{ \cdot - } c{k_{{\rm{TBA,SO}}_4^{ \cdot - }}} c{k_{{\rm{DCF,SO}}_4^{ \cdot - }}} {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } c{k_{{\rm{TBA,SO}}_4^{ \cdot - }}} c{k_{{\rm{MeOH,SO}}_4^{ \cdot - }}} {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_5^{ \cdot - } {\rm{SO}}_4^{ \cdot - } {\rm{SO}}_5^{ \cdot - } {\rm{SO}}_4^{ \cdot - } 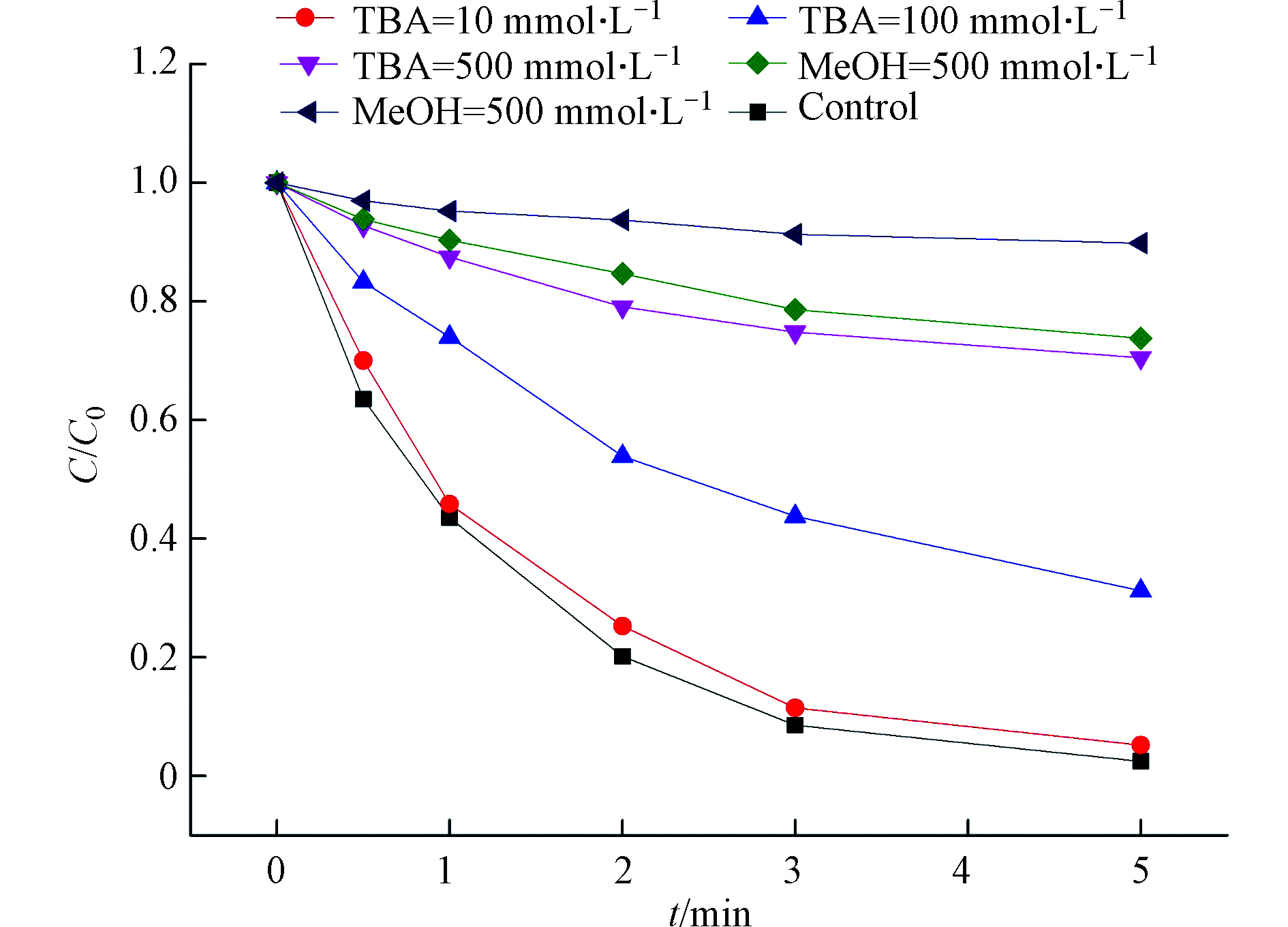 图 9 醇淬灭剂对DCF降解的影响Figure 9. Effect of TBA and MeOH on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, [BS] = 0.2 mmol·L−1, T = 25 ℃.
图 9 醇淬灭剂对DCF降解的影响Figure 9. Effect of TBA and MeOH on DCF degradation in Fe(Ⅲ)/BS system.pH =4.0, [DCF] = [Fe3+] = 10 μmol·L−1, [BS] = 0.2 mmol·L−1, T = 25 ℃.2.5 DCF的降解机理
在Fe(Ⅲ)/BS降解DCF的过程中,共检测到4种转化产物,如表2所示。基于此提出了DCF在该体系中可能的反应机理,主要包括:(1)羟基化反应、(2)脱羧基反应、(3)脱氯氢化反应、(4)甲酰化反应,如图10所示。DCF可通过途径(1)、(2)和(3)分别生成产物m/z 312、252和262,产物m/z 252 通过途径(4)进一步转化为产物m/z 266。
表 2 DCF主要降解产物Table 2. By-products of BPA degradation序号Serial number 荷质比Mass to charge ratio(m/z) 停留时间Retention time/min 分子式Formular 推测结构Proposed structure DCF 296 5.87 C14H11Cl2NO2 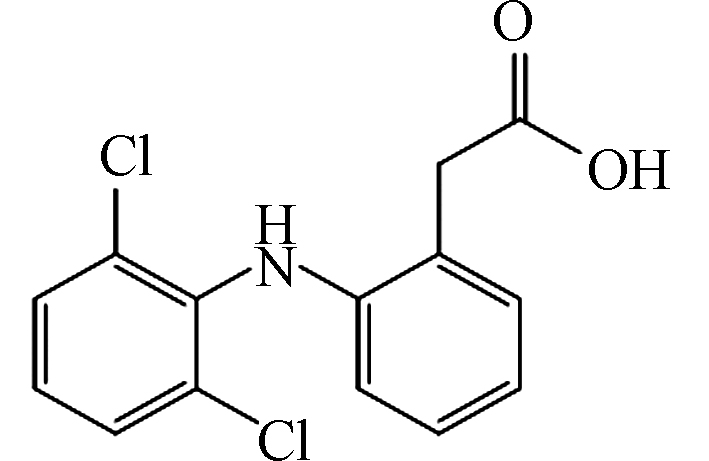
1 312 5.99 C14H1Cl2NO3 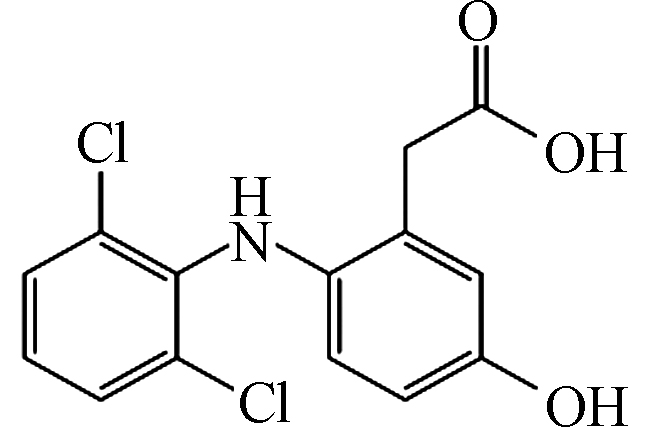
2 266 4.78 C13H9Cl2NO 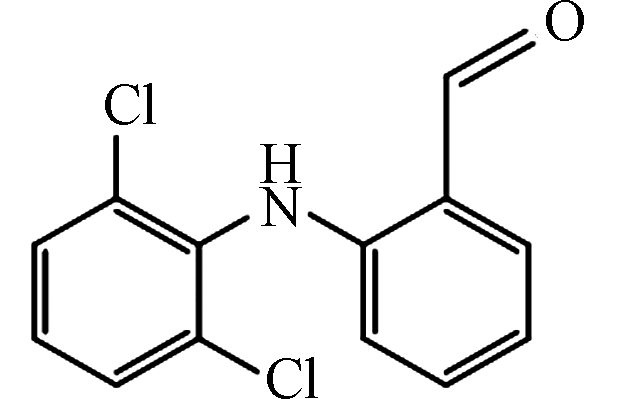
3 262 5.54 C14H12ClNO2 
4 252 5.87 C13H11Cl2N 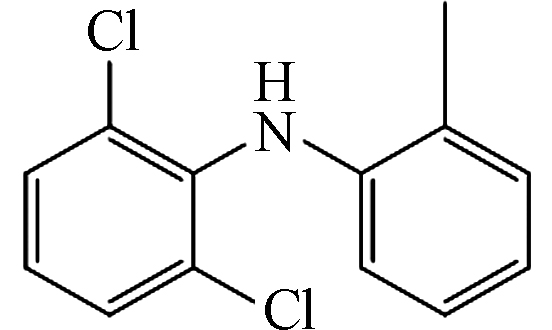 | Show TableDownLoad:
CSV
| Show TableDownLoad:
CSV
3. 结论(Conclusion)
(1)Fe(Ⅲ)/BS可实现Fe(Ⅱ/Ⅲ)自循环,快速高效地降解DCF,其降解规律符合伪一级反应动力学,表观速率常数为0.7453 s−1;表观反应活化能为104.80 kJ·mol−1。
(2)初始pH为4.0时,DCF的降解效率最佳;其降解随着Fe(Ⅲ)和BS的用量增加而增加,然而过量的Fe(Ⅲ)或BS可抑制DCF降解。
(3)DCF在Fe(Ⅲ)/BS的降解与溶解氧浓度无关,在自然搅拌、曝空气或氮气的条件下均可有效的降解;Cl−和FA对DCF的降解起到抑制作用,且两者浓度越高,抑制作用越明显。
(4)Fe(Ⅲ)/BS体系中可能存在
{\rm{SO}}_4^{ \cdot - } {\rm{SO}}_5^{ \cdot - } {\rm{SO}}_4^{ \cdot - } -
图 1 DCF在不同体系的降解(a),Fe(Ⅲ)/BS中Fe和BS浓度变化(b)
Figure 1. Degradation of DCF in different systems (a) and the variation of Fe(Ⅱ), Fe(Ⅲ) and S(Ⅳ) concentrations in Fe(III)/BS system (b).
图 2 pH对DCF降解的影响(a),反应过程中pH的变化情况(b)
Figure 2. Effect of pH (a) and changes in pH (b) on DCF degradation by Fe(Ⅲ)/BS.
图 3 Fe(Ⅲ)浓度对DCF降解的影响
Figure 3. Effect of Fe(Ⅲ) dosage on DCF degradation in Fe(Ⅲ)/BS system.
图 4 BS浓度对DCF降解的影响
Figure 4. Effect of BS dosage on DCF degradation in Fe(Ⅲ)/BS system.
图 5 DCF初始浓度对其自身降解的影响
Figure 5. Effect of initial concentration of DCF on its own degradation in Fe(Ⅲ)/BS system.
图 6 溶解氧的影响
Figure 6. Effect of dissolved oxygen on DCF degradation in Fe(Ⅲ)/BS system.
图 8 氯离子(a)和黄腐酸(b)对DCF降解的影响
Figure 8. Effect of Cl− (a) and FA (b) on DCF degradation in Fe(Ⅲ)/BS system.
图 9 醇淬灭剂对DCF降解的影响
Figure 9. Effect of TBA and MeOH on DCF degradation in Fe(Ⅲ)/BS system.
表 1 目标物与反应自由基的ck值
Table 1. The ck values of target and reactive radical
目标物Reactive species 浓度/(mol·L−1)Concentration HO• {\rm{SO}}_4^{ \cdot - } c/ (L·mol−1·s−1) ck /s−1 k /(L·mol−1·s−1) ck /s−1 DCF 1.0 × 10−5 7.5 × 109 7.5 × 104 9.2 × 109 9.2 × 104 TBA 1.0 × 10−2 7.6 × 108 7.6 × 106 4.0 × 105 4.0 × 103 1.0 × 10−1 7.6 × 107 4.0 × 104 0.5 3.8 × 108 2.0 × 105 MeOH 1.0 × 10−1 9.7 × 108 9.7 × 107 2.5 × 107 2.5 × 106 0.5 4.8 × 108 1.3 × 107
下载: 导出CSV
表 2 DCF主要降解产物
Table 2. By-products of BPA degradation
序号Serial number 荷质比Mass to charge ratio(m/z) 停留时间Retention time/min 分子式Formular 推测结构Proposed structure DCF 296 5.87 C14H11Cl2NO2 </td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">停留时间Retention time/min</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">分子式Formular</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">推测结构Proposed structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:48pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">296</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">C<sub>14</sub>H<sub>11</sub>Cl<sub>2</sub>NO<sub>2</sub></td> <td class="table_top_border2" style="height:48pt;" align="center"><styled-content style="width:3cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" style="height:48pt;" align="center" valign="middle">序号Serial number</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">荷质比Mass to charge ratio(m/z)</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">停留时间Retention time/min</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">分子式Formular</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">推测结构Proposed structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:48pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">296</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">C<sub>14</sub>H<sub>11</sub>Cl<sub>2</sub>NO<sub>2</sub></td> <td class="table_top_border2" style="height:48pt;" align="center"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T1.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">1</td> <td align="center" valign="middle" style="height:48pt;">312</td> <td align="center" valign="middle" style="height:48pt;">5.99</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>1</sub>Cl<sub>2</sub>NO<sub>3</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T2.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">2</td> <td align="center" valign="middle" style="height:48pt;">266</td> <td align="center" valign="middle" style="height:48pt;">4.78</td> <td align="center" valign="middle" style="height:48pt;">C<sub>13</sub>H<sub>9</sub>Cl<sub>2</sub>NO</td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T3.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">3</td> <td align="center" valign="middle" style="height:48pt;">262</td> <td align="center" valign="middle" style="height:48pt;">5.54</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>12</sub>ClNO<sub>2</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T4.jpg"></styled-content></td> </tr><tr><td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">4</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">252</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">C<sub>13</sub>H<sub>11</sub>Cl<sub>2</sub>N</td> <td class="table_bottom_border" style="height:48pt;" align="center"><styled-content style="width:2.5cm"><img class="graphic" src="2020093004-Tab2-T5.jpg"></styled-content></td> </tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">1</td> <td align="center" valign="middle" style="height:48pt;">312</td> <td align="center" valign="middle" style="height:48pt;">5.99</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>1</sub>Cl<sub>2</sub>NO<sub>3</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" style="height:48pt;" align="center" valign="middle">序号Serial number</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">荷质比Mass to charge ratio(m/z)</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">停留时间Retention time/min</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">分子式Formular</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">推测结构Proposed structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:48pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">296</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">C<sub>14</sub>H<sub>11</sub>Cl<sub>2</sub>NO<sub>2</sub></td> <td class="table_top_border2" style="height:48pt;" align="center"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T1.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">1</td> <td align="center" valign="middle" style="height:48pt;">312</td> <td align="center" valign="middle" style="height:48pt;">5.99</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>1</sub>Cl<sub>2</sub>NO<sub>3</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T2.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">2</td> <td align="center" valign="middle" style="height:48pt;">266</td> <td align="center" valign="middle" style="height:48pt;">4.78</td> <td align="center" valign="middle" style="height:48pt;">C<sub>13</sub>H<sub>9</sub>Cl<sub>2</sub>NO</td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T3.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">3</td> <td align="center" valign="middle" style="height:48pt;">262</td> <td align="center" valign="middle" style="height:48pt;">5.54</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>12</sub>ClNO<sub>2</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T4.jpg"></styled-content></td> </tr><tr><td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">4</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">252</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">C<sub>13</sub>H<sub>11</sub>Cl<sub>2</sub>N</td> <td class="table_bottom_border" style="height:48pt;" align="center"><styled-content style="width:2.5cm"><img class="graphic" src="2020093004-Tab2-T5.jpg"></styled-content></td> </tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">2</td> <td align="center" valign="middle" style="height:48pt;">266</td> <td align="center" valign="middle" style="height:48pt;">4.78</td> <td align="center" valign="middle" style="height:48pt;">C<sub>13</sub>H<sub>9</sub>Cl<sub>2</sub>NO</td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" style="height:48pt;" align="center" valign="middle">序号Serial number</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">荷质比Mass to charge ratio(m/z)</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">停留时间Retention time/min</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">分子式Formular</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">推测结构Proposed structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:48pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">296</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">C<sub>14</sub>H<sub>11</sub>Cl<sub>2</sub>NO<sub>2</sub></td> <td class="table_top_border2" style="height:48pt;" align="center"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T1.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">1</td> <td align="center" valign="middle" style="height:48pt;">312</td> <td align="center" valign="middle" style="height:48pt;">5.99</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>1</sub>Cl<sub>2</sub>NO<sub>3</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T2.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">2</td> <td align="center" valign="middle" style="height:48pt;">266</td> <td align="center" valign="middle" style="height:48pt;">4.78</td> <td align="center" valign="middle" style="height:48pt;">C<sub>13</sub>H<sub>9</sub>Cl<sub>2</sub>NO</td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T3.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">3</td> <td align="center" valign="middle" style="height:48pt;">262</td> <td align="center" valign="middle" style="height:48pt;">5.54</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>12</sub>ClNO<sub>2</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T4.jpg"></styled-content></td> </tr><tr><td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">4</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">252</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">C<sub>13</sub>H<sub>11</sub>Cl<sub>2</sub>N</td> <td class="table_bottom_border" style="height:48pt;" align="center"><styled-content style="width:2.5cm"><img class="graphic" src="2020093004-Tab2-T5.jpg"></styled-content></td> </tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">3</td> <td align="center" valign="middle" style="height:48pt;">262</td> <td align="center" valign="middle" style="height:48pt;">5.54</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>12</sub>ClNO<sub>2</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" style="height:48pt;" align="center" valign="middle">序号Serial number</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">荷质比Mass to charge ratio(m/z)</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">停留时间Retention time/min</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">分子式Formular</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">推测结构Proposed structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:48pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">296</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">C<sub>14</sub>H<sub>11</sub>Cl<sub>2</sub>NO<sub>2</sub></td> <td class="table_top_border2" style="height:48pt;" align="center"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T1.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">1</td> <td align="center" valign="middle" style="height:48pt;">312</td> <td align="center" valign="middle" style="height:48pt;">5.99</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>1</sub>Cl<sub>2</sub>NO<sub>3</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T2.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">2</td> <td align="center" valign="middle" style="height:48pt;">266</td> <td align="center" valign="middle" style="height:48pt;">4.78</td> <td align="center" valign="middle" style="height:48pt;">C<sub>13</sub>H<sub>9</sub>Cl<sub>2</sub>NO</td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T3.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">3</td> <td align="center" valign="middle" style="height:48pt;">262</td> <td align="center" valign="middle" style="height:48pt;">5.54</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>12</sub>ClNO<sub>2</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T4.jpg"></styled-content></td> </tr><tr><td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">4</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">252</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">C<sub>13</sub>H<sub>11</sub>Cl<sub>2</sub>N</td> <td class="table_bottom_border" style="height:48pt;" align="center"><styled-content style="width:2.5cm"><img class="graphic" src="2020093004-Tab2-T5.jpg"></styled-content></td> </tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td> </tr><tr><td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">4</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">252</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">C<sub>13</sub>H<sub>11</sub>Cl<sub>2</sub>N</td> <td class="table_bottom_border" style="height:48pt;" align="center"><styled-content style="width:2.5cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" style="height:48pt;" align="center" valign="middle">序号Serial number</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">荷质比Mass to charge ratio(m/z)</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">停留时间Retention time/min</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">分子式Formular</td><td class="table_top_border" style="height:48pt;" align="center" valign="middle">推测结构Proposed structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" style="height:48pt;" align="center" valign="middle">DCF</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">296</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_top_border2" style="height:48pt;" align="center" valign="middle">C<sub>14</sub>H<sub>11</sub>Cl<sub>2</sub>NO<sub>2</sub></td> <td class="table_top_border2" style="height:48pt;" align="center"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T1.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">1</td> <td align="center" valign="middle" style="height:48pt;">312</td> <td align="center" valign="middle" style="height:48pt;">5.99</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>1</sub>Cl<sub>2</sub>NO<sub>3</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T2.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">2</td> <td align="center" valign="middle" style="height:48pt;">266</td> <td align="center" valign="middle" style="height:48pt;">4.78</td> <td align="center" valign="middle" style="height:48pt;">C<sub>13</sub>H<sub>9</sub>Cl<sub>2</sub>NO</td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T3.jpg"></styled-content></td> </tr><tr><td align="center" valign="middle" style="height:48pt;">3</td> <td align="center" valign="middle" style="height:48pt;">262</td> <td align="center" valign="middle" style="height:48pt;">5.54</td> <td align="center" valign="middle" style="height:48pt;">C<sub>14</sub>H<sub>12</sub>ClNO<sub>2</sub></td> <td align="center" style="height:48pt;"><styled-content style="width:3cm"><img class="graphic" src="2020093004-Tab2-T4.jpg"></styled-content></td> </tr><tr><td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">4</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">252</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">5.87</td> <td class="table_bottom_border" style="height:48pt;" align="center" valign="middle">C<sub>13</sub>H<sub>11</sub>Cl<sub>2</sub>N</td> <td class="table_bottom_border" style="height:48pt;" align="center"><styled-content style="width:2.5cm"><img class="graphic" src="2020093004-Tab2-T5.jpg"></styled-content></td> </tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td> </tr></tbody>
</table></div></foreignObject></svg>)
1 312 5.99 C14H1Cl2NO3 2 266 4.78 C13H9Cl2NO 3 262 5.54 C14H12ClNO2 4 252 5.87 C13H11Cl2N
下载: 导出CSV
-
[1] CHEN W, XU J, LU S, et al. Fates and transport of PPCPs in soil receiving reclaimed water irrigation [J]. Chemosphere, 2013, 93(10): 2621-2630. doi: 10.1016/j.chemosphere.2013.09.088 [2] KOSTICH M S, BATT A L, LAZORCHAK J M. Concentrations of prioritized pharmaceuticals in effluents from 50 large wastewater treatment plants in the US and implications for risk estimation [J]. Environmental Pollution, 2014, 184: 354-359. doi: 10.1016/j.envpol.2013.09.013 [3] HALLING S B, NIELESEN S N, LANZKY P, et al. Occurrence fate and effects of pharmaceutical substances in the environment-A review [J]. Chemosphere, 1998, 36(1): 357-393. [4] EBELE A J, ABDALLAH M A, HARRAD S. Pharmaceuticals and personal care products (PPCPs) in the freshwater aquatic environment [J]. Emerging Contaminants, 2017, 3: 1-16. doi: 10.1016/j.emcon.2016.12.004 [5] OAKS J L, GILBERT M, VIRANI M Z, et al. Diclofenac residues as the cause of vulture population decline in Pakistan [J]. Nature, 2004, 427(6975): 630-633. doi: 10.1038/nature02317 [6] CLEUVERS M. Aquatic ecotoxicity of pharmaceuticals including the assessment of combination effects [J]. Toxicology Letters, 2003, 142(3): 185-194. doi: 10.1016/S0378-4274(03)00068-7 [7] LETZEL M, METZNER G, LETZEL T. Exposure assessment of the pharmaceutical diclofenac based on long-term measurements of the aquatic input [J]. Environment International, 2009, 35(2): 363-368. doi: 10.1016/j.envint.2008.09.002 [8] ALVAIINO T, SUAREZ S, KATSOU E, et al. Removal of PPCPs from the sludge supernatant in a one stage nitritation/anammox process [J]. Water Research, 2015, 68: 701-709. doi: 10.1016/j.watres.2014.10.055 [9] ALVARINO T, SUAREZ S, LEMA J M, et al. Understanding the removal mechanisms of PPCPs and the influence of main technological parameters in anaerobic UASB and aerobic CAS reactors [J]. Journal of Hazardous Materials, 2014, 278: 506-513. doi: 10.1016/j.jhazmat.2014.06.031 [10] LIU F F, ZHAO J, WANG S, et al. Effects of solution chemistry on adsorption of selected pharmaceuticals and personal care products (PPCPs) by graphenes and carbon nanotubes [J]. Environmental Science & Technology, 2014, 48(22): 13197-13206. [11] KATSUKI K, HIROE H, YOSHIMASA W. Elimination of Selected Acidic Pharmaceuticals from Municipal Wastewater by an Activated Sludge System and Membrane Bioreactors [J]. Environmental Science and Technology, 2007, 41(10): 3708-3714. doi: 10.1021/es061684z [12] QUINTANA B J, WEISS S, REEMTSMA T. Pathways and metabolites of microbial degradation of selected acidic pharmaceutical and their occurrence in municipal wastewater treated by a membrane bioreactor [J]. Water Research, 2005, 39(12): 2654-2664. doi: 10.1016/j.watres.2005.04.068 [13] BAE S, KIM D, LEE W. Degradation of diclofenac by pyrite catalyzed Fenton oxidation [J]. Applied Catalysis B:Environmental, 2013, 134-135: 93-102. doi: 10.1016/j.apcatb.2012.12.031 [14] JUSTO A, GONZALEZ O, ACENA J, et al. Pharmaceuticals and organic pollution mitigation in reclamation osmosis brines by UV/H2O2 and ozone [J]. Journal of Hazardous Materials, 2013, 263: 268-274. doi: 10.1016/j.jhazmat.2013.05.030 [15] KIM I Y, KIM M K, YOON Y, et al. Kinetics and degradation mechanism of clofibric acid and diclofenac in UV photolysis and UV/H2O2 reaction [J]. Desalination and Water Treatment, 2014, 52(31-33): 6211-6218. doi: 10.1080/19443994.2013.817507 [16] RIZZO L, MERIC S, KASSINOS D, et al. Degradation of diclofenac by TiO2 photo-catalysis: UV absorbance kinetics and process evaluation through a set of toxicity bioassays [J]. Water Research, 2009, 43(4): 979-988. doi: 10.1016/j.watres.2008.11.040 [17] AGUILARLIRA G Y, ALVAREZROMERO G A, ZAMORASUAREZ A, et al. New insights on diclofenac electrochemistry using graphite as working electrode [J]. Journal of Electroanalytical Chemistry, 2017, 794: 182-188. doi: 10.1016/j.jelechem.2017.03.050 [18] NADDEO V, BELGIORNO D, RICCO D, et al. Degradation of diclofenac during sonolysis, ozonation and their simultaneous application [J]. Ultrasonics Sonochemistry, 2009, 16(6): 790-794. doi: 10.1016/j.ultsonch.2009.03.003 [19] NADDEO V, BELGIORNO V, KASSINOS D, et al. Ultrasonic degradation, mineralization and detoxification of diclofenac in water: optimization of operating parameters [J]. Ultrasonics Sonochemistry, 2010, 17(1): 179-185. doi: 10.1016/j.ultsonch.2009.04.003 [20] NIE E, YANG M, YANG X Y, et al. Degradation of diclofenac by ultrasonic irradiation: Kinetic studies and degradation pathways [J]. Chemosphere, 2014, 113: 165-170. doi: 10.1016/j.chemosphere.2014.05.031 [21] NISAR J, SAYED M, KHAN F U, et al. Gamma–irradiation induced degradation of diclofenac in aqueous solution: kinetics, role of reactive species and influence of natural water parameters [J]. Journal of Environmental Chemical Engineering, 2016, 4(2): 2573-2584. doi: 10.1016/j.jece.2016.04.034 [22] DEISTER U, WARNECK P. Photooxidation of {\rm{SO}}_3^{2 - } in Aqueous Solution [J]. Journal of Physical Chemistry, 1990, 94: 2191-2198. doi: 10.1021/j100368a084[23] FISCHER M, WAENECK P. Photodecomposition and Photooxidation of Hydrogen Sulfite in Aqueous Solution [J]. Journal of Physical Chemistry, 1996, 100: 15111-15117. doi: 10.1021/jp953236b [24] ZHANG L, CHEN L, XIAO M, et al. Enhanced decolorization of Orange Ⅱ solutions by the Fe(Ⅱ)-sulfite system under xenon lamp irradiation [J]. Industrial & Engineering Chemistry Research, 2013, 52(30): 10089-10094. [25] 袁亚男. 过渡金属活化亚硫酸盐体系氧化有机污染物的研究 [D]. 武汉: 武汉大学, 2018. YUAN Y. Oxidation of organic compounds in the transition metal ions-activated sulfite systems [D]. Wuhan: Wuhan University, 2018 (in Chinese).
[26] 孙波. NaHSO3活化KMnO4快速氧化水中微量有机污染物的效能与机理 [D]. 哈尔滨: 哈尔滨工业大学, 2017. SUN B. Kinetics and mechanisms on the fast degradation of micro-organic contaminants by bisulfite activated permanganate [D]. Harbin: Harbin Institute of Technology, 2017 (in Chinese).
[27] YUAN Y, LOU T, XU J, et al. Enhanced oxidation of aniline using Fe(Ⅲ)-S(Ⅳ) system: Role of different oxysulfur radicals [J]. Chemical Engineering Journal, 2019, 362: 183-189. doi: 10.1016/j.cej.2019.01.010 [28] ZHOU D N, YUAN Y A, YANG S J, et al. Roles of oxysulfur radicals in the oxidation of Acid Orange 7 in the Fe(Ⅲ)-sulfite system [J]. Journal of Sulfur Chemistry, 2015, 36: 373-384. doi: 10.1080/17415993.2015.1028939 [29] XIAO Q, WANG T, YU S, et al. Influence of UV lamp, sulfur(Ⅳ) concentration, and pH on bromate degradation in UV/sulfite systems: Mechanisms and applications [J]. Water Research, 2017, 111: 288-296. doi: 10.1016/j.watres.2017.01.018 [30] CHEN L, PENG X Z, LIU J H, et al. Decolorization of Orange Ⅱ in Aqueous solution by an Fe(Ⅱ)/sulfite system: Replacement of Persulfate [J]. Industrial Engineering Chemistry Research, 2012, 51: 13632-13638. doi: 10.1021/ie3020389 [31] 张立. Fe(Ⅲ)/S(Ⅳ)体系降解四溴双酚A效能及机理研究 [D]. 武汉: 华中科技大学, 2019. ZHANG L. Study on the degradation of tetrabromobisphenol a by Fe(Ⅲ)/S(Ⅳ) system [D]. Wuhan: Huazhong University of Science and Technology, 2019 (in Chinese).
[32] LENTE G, FABIAN L. Kinetics and mechanism of the oxidation of sulfur(Ⅳ) by iron(Ⅲ) at metal ion excess [J]. Journal of the Chemical Society, Dalton Transactions, 2002, 5: 778-784. [33] BUXTION G V, MCGOWAN S, SALMIN G A, et al. A study of the spectra and reactivity of oxysulphur-radical anions involved in the chain oxidation of S(Ⅳ): A pulse and γ-radiolysis study [J]. Atmospheric Environment, 1996, 30(14): 2483-2493. doi: 10.1016/1352-2310(95)00473-4 [34] WARNECK P, ZIAJKA J. Reaction mechanism of the iron(Ⅲ)-catalyzed autoxidation of bisulfite in aqueous solution: steady state description for benzene as radical scavenger [J]. Berichte der Bunsen-Gesellschaft für physikalische Chemie, 1995, 99: 59-65. [35] HERRMANN H, A REESE, R ZELLNER. Time-resolved UV/VIS diode array absorption spectroscopy of {\rm{SO}}_x^{ \cdot - } (x=3, 4, 5) radical anions in aqueous solution [J]. Journal of Molecular Structure, 1995, 348(15): 183-186.[36] GRGICC I, POZNICC M, BIZJAK M. S(Ⅳ) Autoxidation in atmospheric liquid water: The role of Fe(Ⅱ) and the effect of oxalate [J]. Journal of Atmospheric Chemistry, 1999, 33(1): 89-102. doi: 10.1023/A:1006141913681 [37] GRAEDEL T E, WESCHLER C J. Chemistry within aqueous atmospheric aerosols and raindrops [J]. Reviews of Geophysics, 1981, 19(4): 505-39. doi: 10.1029/RG019i004p00505 [38] BUXTON G V, MALONE T N, SALMON G A. Oxidation of glyoxal initiated by OH in-oxygenated aqueous solution [J]. Journal of the Chemical Society, Faraday Transactions, 1997, 93(16): 2889-2891. doi: 10.1039/a701468f [39] MCELROY W J, WAYGOOD S J. Kinetics of the reactions of {\rm{SO}}_4^{ - } radical with{\rm{SO}}_4^{ - } ,{{\rm{S}}_{\rm{2}}}{\rm{O}}_8^{2 - } , H2O, and Fe2+ [J]. Journal of the Chemical Society Faraday Transactions, 1990, 84(14): 2557-2564.[40] XU J, DING W, WU F, et al. Rapid catalytic oxidation of arsenite to arsenate in an iron(III)/sulfite system under visible light [J]. Applied Catalysis B:Environmental, 2016, 186(5): 56-61. [41] DU J, GUO W, WANG H, et al. Hydroxyl radical dominated degradation of aquatic sulfamethoxazole by Fe0/bisulfite/O2: Kinetics, mechanisms, and pathways [J]. Water Research, 2018, 138(1): 323-332. [42] HUIE R E, NETA P. Rate constants for some oxidations of S(IV) by radicals in aqueous solutions [J]. Atmospheric Environment, 1987, 21(8): 1743-1747. doi: 10.1016/0004-6981(87)90113-2 [43] HUIE R E, CLIFTON C L, ALTSTEIN N. A pulse radiolysis and flash photolysis study of the radicals {\rm{SO}}_2^{ \cdot - } ,{\rm{SO}}_3^{ \cdot - } ,{\rm{SO}}_4^{ \cdot - } and{\rm{SO}}_5^{ \cdot - } [J]. Radiation Physics and Chemistry, 1989, 33(4): 361-370.[44] HUSS A, LIM P. K, Eckert C. A. Oxidation of aqueous sulfur dioxide. 2. High-pressure studies and proposed reaction mechanisms [J]. Journal of Physical Chemistry, 1982, 86: 4229-4233. doi: 10.1021/j100218a028 [45] WANG, H, WANG S, LIU Y, et al. Degradation of diclofenac by Fe(Ⅱ)-activated bisulfite: Kinetics, mechanism and transformation products [J]. Chemosphere, 2019, 237: 124518. doi: 10.1016/j.chemosphere.2019.124518 [46] NETA P, HUIE R E, ROSS A B, Rate Constants for reactions of inorganic radicals in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17: 1027-1284. [47] ANIPSITAKIS G P, DIONYSIOU D D. Radical generation by the interaction of transition metals with common oxidants [J]. Environmental Science and Technology, 2004, 38: 3705-3712. doi: 10.1021/es035121o [48] 邹景. 羟胺对Fe2+/过硫酸盐体系的强化效能与机理研究 [D]. 哈尔滨: 哈尔滨工业大学, 2016. ZOU J. Enhanced effectiveness and mechanism of Fe2+/persulfate system with hydroxylamine[D]. Harbin: Harbin Institute of Technology, 2016 (in Chinese).
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 5376
- HTML全文浏览数: 5376
- PDF下载数: 113
- 施引文献: 0


























































