













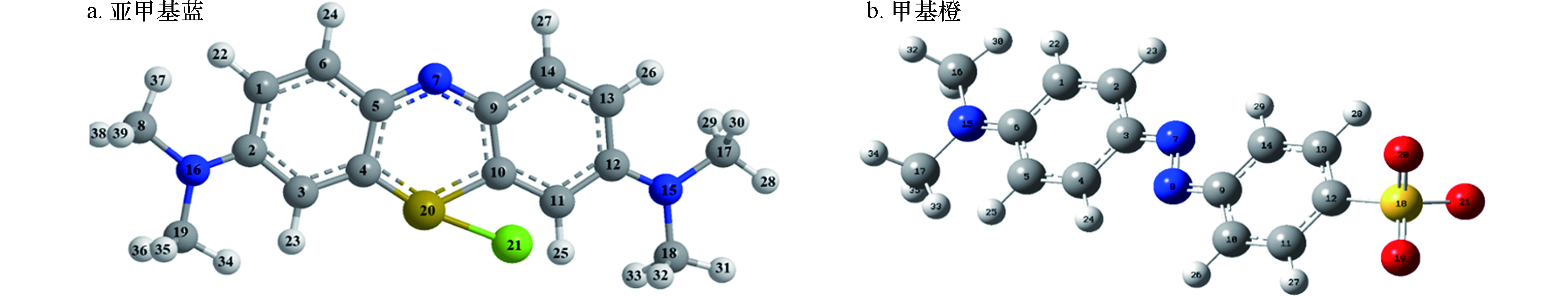















-
高级氧化法(AOPs)目前广泛应用于不同废水的处理[1],并且基于原位生成的具有强氧化性的活性物种,如羟基自由基(OH·)等,能几乎将所有类型的有机污染物降解[2]。尽管OH·氧化电位高(E0=2.7 V),能氧化大部分有机化合物,甚至可以将其完全矿化为CO2和H2O[3-5],但OH·降解有机污染物的过程是多步反应,通常需要酸性条件,受pH的影响较大[6]。此外,在含有大量溶解有机物(DOMs)和碳酸盐/重碳酸盐阴离子的复杂环境基质中,OH·的效率受到严重限制[7],在水中的使用寿命不长,与有机污染物发生反应的概率也较低[8]。
近年来,以硫酸根自由基(
${\rm{SO}}_4^ - \cdot $ )为基础的AOPs针对有机污染物有着较高的反应活性,面对复杂的环境基质也具有高选择性[9]。${\rm{SO}}_4^ - \cdot $ (E0=2.5—3.1 V)比OH·(E0=1.8—2.7 V)具有更高的氧化还原电位[10],因此${\rm{SO}}_4^ - \cdot $ 能够高效的氧化有机污染物。OH·受有机物水体的pH影响较大,即去除效率随着pH的增加而降低,而${\rm{SO}}_4^ - \cdot $ 反应活性却具有较宽的pH范围[7];${\rm{SO}}_4^ - \cdot $ 在水中的存在寿命更长(3×10−5—4×10−5 s)且更加稳定,与有机污染物发生反应的概率也高于OH·(2×10−8 s)[8];${\rm{SO}}_4^ - \cdot $ 的自清除作用不明显,可以在水中产生高浓度的自由基,进而改善了高浓度废水的反应动力学,呈现出高反应的化学计量效率。${\rm{SO}}_4^ - \cdot $ 是通过前体分子的激活而产生的。已知常用的前体有过一硫酸盐(PMS,${\rm{HSO}}_5^ - $ )和过硫酸盐(PS,${{\rm{S}}_2}{\rm{O}}_8^{2 - } $ )[11]。活化方法多样,如加入金属催化剂Fe、Cu、Co[12]、提高温度(热活化)[13]、紫外线照射或超声波活化[14]等方法。这些方法广泛应用于处理难降解有机物,如内分泌干扰物(EDC)、医药和个人护理产品和藻毒素[15]。在活化过硫酸盐体系中,主导自由基(${\rm{SO}}_4^ - \cdot $ 或OH·)的类型在很大程度上取决于工艺条件、废水组成和使用的活化方法。在一些研究中,通过自由基猝灭实验调查了体系中的起主导作用的活性物种。然而,在活化过硫酸盐体系中的自由基与不同类型有机物的相互作用仍不清楚,仍需进一步解决。在以往的研究和应用中,过硫酸盐主要由紫外、热、过渡金属离子等方法激活[16],Liu[17]等通过热活化过硫酸盐体系降解布洛芬的研究,发现反应温度对污染物降解起到关键作用,且体系中硫酸根自由基对降解布洛芬起主导作用,布洛芬的降解随着体系温度的升高而增加。在探究光活化过硫酸盐体系对聚乙烯醇的降解实验中,对比了254 nm和365 nm两个波长的去除率,发现在254 nm时,不到5 min的去除率达到了100%,而在365 nm下,30 min的去除率只有93%[18]。在紫外活化过硫酸盐体系中,波长和紫外辐射率对过硫酸盐的激活和有机物降解起着重要作用,为了缩短反应时间,提高反应效率,波长254 nm在紫外活化体系中比较常用[19]。虽然紫外光活化方法是一种经济有效的方法,甚至可以代替热活化,但由于紫外线穿透水的能力有限,所以这种方法也受到一定的限制[20]。探究连续添加Fe2+活化过硫酸盐的方法来去除有机污染物的情况,经过4次连续的添加使双酚A的降解率从49%提高到97%,连续添加Fe2+最终去除率可达到100%[21]。在基于过硫酸盐的AOPs中,
$ {\rm{SO}}_4^ - \cdot $ 自由基能否在不同的水基质中高效、快速地被激活是该方法的应用关键。而目前对于各种激活模式的系统性比较研究存在一定的不足,对降解途径和不同阴离子对系统的影响的研究也比较缺乏,机理分析不够深入,所以不同活化过硫酸盐体系的系统性比较及相关机理的探究是尤为必要的。本研究通过比较3种不同的活化过硫酸盐体系对两种染料的降解效果,进而研究不同活化方法的降解效率及反应机理。基于
${\rm{SO}}_4^ - \cdot $ 的选择性,选择了两种具有代表性的染料(阳离子染料亚甲基蓝和阴离子染料甲基橙)为目标污染物。通过自由基捕获实验,判断不同活化过硫酸盐体系对染料降解起到主要作用的活性物种。通过LC-MS测定及分子轨道计算,探究两种染料在活化过硫酸盐体系中的降解路径及中间产物。通过探索4种阴离子(${\rm{HCO}}_3^ - $ 、${\rm{NO}}_3^ - $ 、Cl−、Br−)对3种活化过硫酸盐体系降解染料的影响,阐明了基于${\rm{SO}}_4^ - \cdot $ 的AOPs在实际废水处理中的应用前景。 -
过硫酸钠来自上海安普斯科技有限公司,硫酸亚铁(AR)来自美佳纳米材料,氯化钠、溴化钠、硝酸钠、碳酸氢钠、甲醇、叔丁醇、亚甲基蓝(MB)、甲基橙(MO)均来自上海安谱科技有限公司。所有化学品均为分析纯,没有经过进一步的净化。所有溶液均使用去离子水进行配制。
-
热-Na2S2O8体系对MB、MO的降解,将10 mg·L−1的MB与MO溶液分别于锥形瓶中,快速加入一定浓度(8 mmol·L−1)的过硫酸钠溶液,均匀混合使之充分反应,本次实验采用电热恒温水浴锅控温(60 ℃);向体系中投加过量的甲醇(MA)与叔丁醇(TBA),对比MB、MO的降解情况,由于体系反应迅速,此次实验将Na2S2O8浓度调节为4.0 mmol·L−1;将MB、MO溶液(10 mg·L−1)、过硫酸钠溶液(8 mmol·L−1)于锥形瓶中充分混合,分别添加不同浓度的无机阴离子(
${\rm{HCO}}_3^ - $ 、${\rm{NO}}_3^ - $ 、Cl−、Br−)置入水浴恒温震荡锅中进行反应,对比热-Na2S2O8体系对MB、MO的降解情况。光-Na2S2O8体系对MB、MO的降解,将10 mg·L−1的MB与MO溶液分别置于自制耐热光反应器中,快速加入一定浓度(8 mmol·L−1)的过硫酸钠溶液,本实验光活化反应容器为亚克力材料制作而成,灯源用以模拟太阳光照,型号为HPI-T Plus 250 w的金属卤化灯,以石英冷肼为循环水装置,冷却系统为KDC低温恒温槽。向体系中投加过量的甲醇(MA)与叔丁醇(TBA),对比MB、MO的降解情况;将MB、MO溶液(10 mg·L−1)、过硫酸钠溶液(8 mmol·L−1) 于锥形瓶中充分混合,分别添加不同浓度的无机阴离子(
${\rm{HCO}}_3^ - $ 、${\rm{NO}}_3^ - $ 、Cl−、Br−)置入自制耐热光反应器中进行反应,对比光-Na2S2O8体系对MB、MO的降解情况。Fe2+-Na2S2O8体系对MB、MO的降解,将10 mg·L−1的MB与MO溶液分别加入一定浓度的硫酸亚铁溶液(0.3 mmol·L−1)与过硫酸钠溶液(1.2 mmol·L−1)于锥形瓶中,均匀混合使之充分反应,冷却系统为KDC低温恒温槽;向体系中投加过量的甲醇(MA)与叔丁醇(TBA),对比MB、MO的降解情况;将MB、MO溶液(10 mg·L−1)、过硫酸钠溶液(1.2 mmol·L−1)与硫酸亚铁溶液(0.3 mmol·L−1)充分混合,分别添加不同浓度的无机阴离子(
${\rm{HCO}}_3^ - $ 、${\rm{NO}}_3^ - $ 、Cl−、Br−)进行反应,对比Fe2+-Na2S2O8体系对MB、MO的降解情况。 -
亚甲基蓝(665 nm)与甲基橙(464 nm)均采用可见光分光光度计(722N型)进行测定,制作亚甲基蓝/甲基橙-吸光度标准工作曲线。两种染料浓度可根据测定的吸光度对照标准工作曲线得出。
在热活化过硫酸盐体系降解亚甲基蓝和甲基橙的实验中,取反应中间时间的反应液,进行中间产物分析,推测可能的降解路径。采用超高相液相色谱-质谱联用仪(美国,安捷伦6460)对亚甲基蓝降解的中间产物进行测定,测定条件:离子源为ESI离子源,毛细管电压:3.5 kV,圆锥电压为52 V,喷雾器压力为40 psi,干燥气体流速为800 L·h−1,流动相为乙腈∶甲酸(0.1%)=1∶1。采用超高相液相色谱-质谱联用仪(美国,安捷伦6460)对甲基橙降解的中间产物进行测定,测定条件:离子源为ESI离子源,毛细管电压:3.5 kV;圆锥电压为52 V,喷雾器压力为40 psi,干燥气体流10 L·min−1,流动相为乙腈∶醋酸铵(0.01 mol·L−1)=30∶70。
-
利用Gaussian 09程序进行了分子轨道计算,得到了能量最小的最佳构象。测定了最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的EHOMO2+ELUMO2值。
-
在3种活化过硫酸盐体系中,可观察到在180 min内,除了甲基橙(图1b)在亚铁离子活化过硫酸盐体系中降解率为74.44%,剩余体系两种染料(图1b,图1a)的降解率均达到了100%。表明活化过硫酸盐体系均可高效的降解两种具有代表性的染料。而且在同一体系中,对亚甲基蓝的降解作用要强于对甲基橙的降解作用,这是由于亚甲基蓝为吩噻嗪盐类染料,是一种芳香杂环化合物,属于碱性阳离子染料,正电荷相对不稳定,对体系的变化十分敏感,而甲基橙是一种典型的偶氮染料,属于碱性阴离子染料,其分子中苯胺与苯磺酸钠基团由N=N键相连,属于难生物降解有机物,且甲基橙结构中的键能大于亚甲基蓝的化学键,所以甲基橙的分子结构比亚甲基蓝的分子结构更为稳定[22]。
3种活化过硫酸盐体系各有不同的优劣,Fe2+-Na2S2O8体系过硫酸钠的消耗量最少,且无能量的消耗,但是体系中引入了外来的Fe2+,后期需要将Fe2+去除;heat-Na2S2O8体系中虽然无外来离子的引入,但体系耗能;hv-Na2S2O8体系虽然无外来离子的添加,但是消耗能量最大,且体系反应时间最长,降解染料的速率较慢。所以出于综合经济效益考虑,最佳优化体系为heat-Na2S2O8体系。
-
在过硫酸盐活化体系中可以产生硫酸根自由基(
${\rm{SO}}_4^ - \cdot $ )和羟基自由基(OH·)[23],${\rm{SO}}_4^ - \cdot $ 和OH·与甲醇(MA)的反应速率常数分别为0.9×108—1.3×108 L·(mol ·s)−1和8.0×108—10×108 L·(mol ·s)−1,而OH·与叔丁醇(TBA)的反应速率常数为3.8×108—7.6×108 L·(mol ·s)−1之间,是${\rm{SO}}_4^ - \cdot $ 与叔丁醇(TBA)反应速率常数〔4.0×105—9.1×105 L·(mol ·s)−1〕的1000倍[24]。因此,分别向3种体系中加入过量的MA与TBA,对比3种活化体系中两种染料的降解,进而鉴定体系中的${\rm{SO}}_4^ - \cdot $ 和OH·的贡献作用。由图2可看出,3种体系中MA与TBA的加入均可抑制两种染料的降解。在heat-Na2S2O8体系中,MA与TBA的过量引入,抑制了两种染料的降解,且引入MA的降解抑制效果明显强于引入TBA的降解抑制效果,可说明体系内
${\rm{SO}}_4^ - \cdot $ 和OH·均存在,且${\rm{SO}}_4^ - \cdot $ 占主导作用。这是由于在heat-Na2S2O8体系中,过硫酸盐中的O-O键由于体系中的热辐射作用而发生断裂反应,生成2个分子量的${\rm{SO}}_4^ - \cdot $ [25] (${{\rm{S}}_2}{\rm{O}}_8^{2 - } $ + heat →$2{\rm{SO}}_4^ - \cdot $ ),而过硫酸钠的加热状态相比其他条件下,能够产生较多的${\rm{SO}}_4^ - \cdot $ ,这既归因于过硫酸盐活化效率的提高,也归因于反应速率常数的增加,使${\rm{SO}}_4^ - \cdot $ 的产生变得更容易[26],${\rm{SO}}_4^ - \cdot $ 此时占主要反应自由基,故${\rm{SO}}_4^ - \cdot $ 占主导作用。在hv-Na2S2O8体系中,引入MA的降解抑制效果与引入TBA的降解抑制效果基本持平,过硫酸盐中的O-O键在模拟太阳光的照射下可以产生断裂反应,由于过硫酸盐是对称氧化剂,所以可生成2个分子量的
${\rm{SO}}_4^ - \cdot $ 。在初期光解产生${\rm{SO}}_4^ - \cdot $ 的过程中,${\rm{SO}}_4^ - \cdot $ 此时占主要反应自由基,而随着反应时间的继续,部分${\rm{SO}}_4^ - \cdot $ 在水溶液中可自发的转化为OH· (${\rm{SO}}_4^ - \cdot $ + H2O→${\rm{SO}}_4^ {2-} $ + OH·)[7]。而在pH值提高时,OH·生成更多 (${\rm{SO}}_4^ - \cdot $ + OH−→${\rm{SO}}_4^ {2- } $ + OH·)[7],即OH·此时占主要反应自由基。所以,在hv-Na2S2O8体系降解染料的过程中,${\rm{SO}}_4^ - \cdot $ 和OH·均占主导作用。在Fe2+-Na2S2O8体系中,引入TBA的降解抑制效果明显强于引入MA的降解抑制效果,说明体系中
${\rm{SO}}_4^ - \cdot $ 和OH·均存在,而此时OH·占主导作用。这是由于亚铁离子在室温下除了可以催化过硫酸盐产生强氧化性的${\rm{SO}}_4^ - \cdot $ 外[27],还可与溶液中少量溶解氧反应产生OH·。Na2S2O8水解可生成H2O2,而H2O2与亚铁离子发生芬顿反应可生成OH·。过硫酸盐体系所产生的${\rm{SO}}_4^ - \cdot $ 通过链传递过程也可生成OH· [28]。所以Fe2+-Na2S2O8体系降解染料的过程中,OH·占主导作用。MA与TBA对3种体系降解甲基橙的抑制效果与上述降解亚甲基蓝的抑制效果趋向一致。在在同一体系中,对降解亚甲基蓝的抑制效果要强于甲基橙。这是由于两种捕获剂优先与自由基发生反应,此时染料的种类并不对其产生影响,并且${\rm{SO}}_4^ - \cdot $ 和OH·对降解亚甲基蓝的贡献要大于对甲基橙的贡献,这与上述2.1的现象相吻合。 -
从2.2节可以看出,
${\rm{SO}}_4^ - \cdot $ 和OH·在亚甲基蓝和甲基橙的降解中起到一定的主导作用。由于3种体系的降解中间产物和降解路径基本相同,此时以热活化过硫酸盐体系为代表,探究亚甲基蓝和甲基橙的降解中间产物以及降解路径。在热活化体系中,亚甲基蓝在降解过程中出现了许多小分子物质的峰族(表1,图3(a1—f1),图4),其中的一个降解路径是:大量过硫酸盐中的O-O键由于体系的热辐射作用而发生断裂,生成大量
${\rm{SO}}_4^ - \cdot $ [25],${\rm{SO}}_4^ - \cdot $ 率先攻击EHOMO2+ELUMO2值较大(图5a,表2)的21Cl硫氯键,硫氯键优先断裂[29],生成C12H10N3S分子,进而在${\rm{SO}}_4^ - \cdot $ 的攻击下,噻嗪环开环降解,生成小分子有机物;另一个路径是:${\rm{SO}}_4^ - \cdot $ 攻击EHOMO2+ELUMO2值较大(图5a,表2)的7N碳氮双键,碳氮双键优先断裂,生成C16H23N3SO分子,然后在${\rm{SO}}_4^ - \cdot $ 的攻击下噻嗪环开环降解,生成C8H8N2O2、C8H11NSO3Na分子,进而生成C6H6O3Na分子,随着反应时间的延续,最后在${\rm{SO}}_4^ - \cdot $ 的作用下,生成C2H5NO4Na分子,最终矿化为CO2和H2O。在热活化体系中,甲基橙在降解过程中出现了许多小分子物质的峰族(表3,图3(a2—d2),图6),其中的降解路径为:由于大量过硫酸盐中的O-O键在热辐射作用下发生断裂反应,生成大量
${\rm{SO}}_4^ - \cdot $ [25],${\rm{SO}}_4^ - \cdot $ 率先攻击EHOMO2+ELUMO2值相对较大(图5b,表4)的7N,8N的偶氮键[29],导致发色基团-N=N-键优先断裂[30],随着反应时间的延续生成C6H7NO3S、C8H12N2等小分子,在${\rm{SO}}_4^ - \cdot $ 的攻击下,进一步生成C4H4O4分子,最终矿化为CO2和H2O。热活化体系在降解两种染料的整个过程中,OH·与${\rm{SO}}_4^ - \cdot $ 均存在,但${\rm{SO}}_4^ - \cdot $ 在降解过程中一直起到了主导作用。 -
由于无机阴离子会优先与
${\rm{SO}}_4^ - \cdot $ 发生反应,优先消耗${\rm{SO}}_4^ - \cdot $ ,其反应效率要远远高于${\rm{SO}}_4^ - \cdot $ 对亚甲基蓝(MB)与甲基橙(MO)的降解效率,所以染料的种类在此过程中并不产生影响,故本文选择以MB为代表,如图7所示,探究四种阴离子(${\rm{HCO}}_3^ - $ 、${\rm{NO}}_3^ - $ 、Cl−、Br−)对3种体系降解MB的影响。碳酸氢根离子(
$ {\rm{HCO}}_3^ - $ )均抑制了3种活化反应体系对MB的降解(图7a),其中对Fe2+-Na2S2O8体系的抑制作用最强,其次是hv-Na2S2O8体系,对heat-Na2S2O8体系的抑制作用最弱。对Fe2+-Na2S2O8体系的抑制作用与加入的${\rm{HCO}}_3^ - $ 浓度成正相关,而${\rm{HCO}}_3^ - $ 对heat-Na2S2O8体系与hv-Na2S2O8体系的抑制作用在浓度较低的范围内与${\rm{HCO}}_3^ - $ 浓度呈正相关。这主要是由于在heat-Na2S2O8体系与hv-Na2S2O8体系中,${\rm{HCO}}_3^ - $ 会与${\rm{SO}}_4^ - \cdot $ 发生反应,从而消耗${\rm{SO}}_4^ - \cdot $ [31-32],同时${\rm{HCO}}_3^ - $ 会电离生成${\rm{CO}}_3^ {2- } $ ,而${\rm{CO}}_3^ {2- } $ 同样会竞争${\rm{SO}}_4^ - \cdot $ [32]。随着${\rm{HCO}}_3^ - $ 的引入,会导致体系由酸性变为弱碱性,在弱碱性的条件下,反应液中的氢氧根会消耗部分${\rm{SO}}_4^ - \cdot $ ,也会使得降解率降低。在Fe2+-Na2S2O8体系中,随着${\rm{HCO}}_3^ - $ 浓度的增加,降解作用对阴离子的浓度变化表现较为敏感。${\rm{HCO}}_3^ - $ 的添加会使体系的环境变为弱碱性,即导致体系pH值为6—8。在弱碱性条件下,Fe2+易与OH−反应生成Fe(OH)2,同时${\rm{HCO}}_3^ - $ 与其电离产生的${\rm{CO}}_3^{2 -} $ 在反应体系中同样会与$ {\rm{SO}}_4^ - \cdot $ 发生竞争。而${\rm{HCO}}_3^ - $ 与Fe2+的摩尔比≥1,故${\rm{SO}}_4^ - \cdot $ 将很快被$ {\rm{HCO}}_3^ - $ 与${\rm{CO}}_3^ {2- } $ 反应完全,无新的${\rm{SO}}_4^ - \cdot $ 产生,虽然会产生少量的${\rm{HCO}}_3^ - \cdot $ 和${\rm{CO}}_3^ {2-} \cdot $ ,但由于比较少的产生量且Fe2+大部分被完全反应,故${\rm{HCO}}_3^ - \cdot$ 和${\rm{CO}}_3^ {2-} \cdot $ 对MB的降解作用在整个体系中可忽略不计[33-34]。而heat-Na2S2O8与hv-Na2S2O8的活化方式均属于外加能源,由于过硫酸盐的浓度在两种体系中较高,${\rm{HCO}}_3^ - \cdot $ 和${\rm{CO}}_3^ {2-} \cdot$ 的产生量较大,所以${\rm{HCO}}_3^ {-} \cdot$ 的抑制作用相比于Fe2+-Na2S2O8体系较弱。硝酸根离子(
${\rm{NO}}_3^ - $ )均抑制了3种活化反应体系对MB的降解(图7b),其中对Fe2+-Na2S2O8体系的抑制作用最强,其次是heat-Na2S2O8体系,对hv-Na2S2O8体系的抑制作用最弱。对Fe2+-Na2S2O8体系的抑制作用与加入的${\rm{NO}}_3^ - $ 浓度呈正相关,而${\rm{NO}}_3^ - $ 对heat-Na2S2O8体系与hv-Na2S2O8体系的抑制作用在较低的浓度范围内与${\rm{NO}}_3^ - $ 浓度呈正相关,在较高的浓度范围内表现为负相关。这主要是由于在heat-Na2S2O8体系中,${\rm{NO}}_3^ - $ 会与${\rm{SO}}_4^ - \cdot $ 发生反应,从而消耗${\rm{SO}}_4^ - \cdot $ [31, 35],使得降解率降低。当${\rm{NO}}_3^ - $ 浓度升高时,由于增加的${\rm{NO}}_3^ - $ 会促进${\rm{NO}}_3^ - \cdot $ 自由基的增多,而${\rm{NO}}_3^ - \cdot $ 在体系中具有一定的氧化能力,对MB起到一定程度的降解作用,从而缓解了${\rm{NO}}_3^ - $ 对降解MB的抑制作用,因此 heat-Na2S2O8体系对MB的降解效果又有所升高。当${\rm{NO}}_3^ - $ 浓度进一步增强时,抑制作用随${\rm{NO}}_3^ - $ 浓度升高而增强,这是由于${\rm{NO}}_3^ - $ 消耗的$ {\rm{SO}}_4^ - \cdot $ 多于产生的${\rm{NO}}_3^ - \cdot $ ,或者产生的${\rm{NO}}_3^ - \cdot $ 自由基消耗完全,故随着${\rm{NO}}_3^ - $ 浓度的升高,对heat-Na2S2O8体系降解MB的抑制作用增强。在Fe2+-Na2S2O8体系中,降解反应对${\rm{NO}}_3^ - $ 的浓度变化也是较为敏感,由于Fe2+与${\rm{SO}}_4^ - \cdot $ 的反应速率要低于$ {\rm{NO}}_3^ - $ 与${\rm{SO}}_4^ - \cdot $ 的反应速率,从而影响体系中$ {\rm{SO}}_4^ - \cdot $ 对MB的降解作用[36, 37]。在hv-Na2S2O8体系中,虽然原理与heat-Na2S2O8体系大体一致,但OH·在模拟太阳光的条件会继续产生,故总体上${\rm{NO}}_3^ - $ 对hv-Na2S2O8体系降解MB的抑制作用最弱。氯离子(Cl−)均抑制了3种活化反应体系对MB的降解(图7c),其中对Fe2+-Na2S2O8体系的抑制作用最强,对heat-Na2S2O8体系与hv-Na2S2O8体系的抑制作用较弱且接近,对Fe2+-Na2S2O8体系的抑制作用与加入的Cl−浓度呈正相关。这是由于在heat-Na2S2O8体系与hv-Na2S2O8体系中,Cl−会与
${\rm{SO}}_4^ - \cdot $ 发生反应,从而消耗${\rm{SO}}_4^ - \cdot $ [31, 35],而当Cl−浓度升高时,Cl−产生的Cl·可以一定程度上氧化MB,Cl·与Cl−反应生成的${\rm{Cl}}_2^ - \cdot $ 同样具有一定的氧化能力[38],所以Cl−对heat-Na2S2O8体系与hv-Na2S2O8体系降解MB的抑制作用较弱。在Fe2+-Na2S2O8体系中,降解作用对Cl−的浓度变化较为敏感,由于Fe2+与${\rm{SO}}_4^ - \cdot $ 的反应速率要低于Cl−与${\rm{SO}}_4^ - \cdot $ 的反应速率,从而大大影响了${\rm{SO}}_4^ - \cdot $ 对MB的降解作用[38-40],故抑制作用最为明显。溴离子(Br−)均抑制了3种活化反应体系对MB的降解(图7d),其中对Fe2+-Na2S2O8体系的抑制作用最强,对heat-Na2S2O8体系与hv-Na2S2O8体系的抑制作用较弱且接近,Fe2+-Na2S2O8体系中抑制作用与加入的Br−浓度呈正相关。这是由于在heat-Na2S2O8体系与hv-Na2S2O8体系中,Br−会与
${\rm{SO}}_4^ - \cdot $ 发生反应,从而消耗${\rm{SO}}_4^ - \cdot $ [31, 35],使得降解率降低。随着Br−浓度升高,B·自由基增多,Br·与Br−反应也可生成${\rm{Br}}_2^ - \cdot $ ,而Br·与$ {\rm{Br}}_2^ - \cdot $ 具有一定的氧化能力,对MB起到一定程度的降解作用,从而缓解了Br−对降解MB的抑制作用,故Br−对heat-Na2S2O8体系与hv-Na2S2O8体系的抑制作用较弱。在Fe2+-Na2S2O8体系中,降解作用对Br−的浓度变化较为敏感,由于Fe2+与${\rm{Br}}_2^ - \cdot $ 反应速率要低于Br−与${\rm{Br}}_2^ - \cdot $ 的反应速率,从而大大影响了${\rm{Br}}_2^ - \cdot $ 对MB的降解作用[41-42],故抑制作用最为明显。加入的无机阴离子基本上都是对Fe2+-Na2S2O8体系降解MB的抑制作用最强,这是由于加入的无机阴离子与亚铁离子竞争
${\rm{Br}}_2^ - \cdot $ ,且${\rm{Br}}_2^ - \cdot $ 在Fe2+-Na2S2O8体系中对无机阴离子的反应能力相比较其他体系更强[43]。此外,亚铁离子对于pH的变化也相对敏感,尤其当pH> 3时,亚铁离子倾向于共沉淀,因此也会削弱Fe2+-Na2S2O8体系的氧化作用[27],由此本部分也表明heat-Na2S2O8体系的适用性更强。在本文各个实验探究中,除${\rm{HCO}}_3^ - $ 对过硫酸盐体系的pH值有缓冲作用致使pH值为6—8外,其他部分三个体系的初始pH值均为2—3。这是由于在保持非人工调节pH的情况下,初始加入的过硫酸盐所导致的pH值为2—3。并且追踪体系的整个反应过程的pH值,发现pH变化的影响较小。 -
在本研究中,发现了3种活化过硫酸盐高级氧化技术均能高效的降解两种具代表性的有机染料(MB、MO),且对亚甲基蓝的降解效率要好于对甲基橙的降解效率。3种反应体系各有优劣,但基于综合经济性考虑,热活化过硫酸盐体系可作为最佳优化体系。在以后实际应用中可以考虑在热活化过硫酸盐高级氧化技术的基础上,增加加热时间以及药剂投加量等优化方式来进行实际废水处理。
在3种体系中,叔丁醇和甲醇的加入均可抑制两种染料的降解,并且对亚铁离子活化过硫酸盐体系降解染料的抑制作用最强,这与体系中产生的羟基自由基和硫酸根自由基有着很大的关系,这两种自由基在3种体系对染料降解过程中有着不同的贡献。本研究正是通过对活性物质的鉴定,进一步了解了降解机制,证实了产生的主导自由基(
${\rm{SO}}_4^ - \cdot $ 或OH·)的类型在很大程度上可能取决于工艺条件、废水组成和使用的活化方法等。3种活化过硫酸盐体系的中间产物和降解路径基本相同,并以热活化过硫酸盐体系为代表,阐明了亚甲基蓝与甲基橙的中间产物以及降解路径。当外界条件发生变化时,比如温度、溶剂和压力等,亚甲基蓝会有分子聚集现象产生,而甲基橙分子结构中苯磺酸钠基团与苯胺由氮氮双键相连,分子结构非常稳定,属于难生物降解的有机污染物范畴。本文的研究结果可为两种可具代表性的染料废水的高效处理提供一定参考。最后,4种阴离子(
${\rm{HCO}}_3^ - $ 、${\rm{NO}}_3^ - $ 、Cl−、Br−)对 3种活化过硫酸盐体系降解两种染料均有一定的抑制作用,尤其在亚铁离子活化过硫酸盐体系中对四种阴离子的浓度变化更为敏感,通过机理分析证实了热活化过硫酸盐高级氧化技术在实际废水处理中有着广阔的应用前景。
不同活化过硫酸盐体系的机理分析及不同无机阴离子的作用:以两种有机染料为例
Mechanism analysis of different activated persulfate systems and effects of different inorganic anions: a case study of two organic dyes
-
摘要: 高级氧化技术(AOPs)广泛应用于不同的废水处理,并目前此类技术多以羟基自由基(OH·)的产生为主。近年来,基于硫酸根自由基(
${{\rm{SO}}_4^ - \cdot} $ )的AOPs因其对有机污染物的高反应活性和对复杂环境基质的高选择性而备受关注。但是,在对各种活化方式的系统比较方面还存在一些不足,对降解途径和不同阴离子对体系的影响也缺乏研究。本文通过两种染料的降解对3种活化过硫酸盐体系进行了系统性的比较,并结合自由基捕获实验(甲醇,叔丁醇)和产物分析,研究了体系降解机制及路径。研究了4种阴离子(${ {\rm{HCO}}_3^ - }$ 、${{\rm{NO}}_3^ - }$ 、Cl−、Br−)对3种不同活化过硫酸盐体系降解染料的影响。结果表明,${{\rm{SO}}_4^ - \cdot }$ 与OH·在3种活化体系中对染料降解均有贡献。在热活化体系(heat-Na2S2O8)中,对染料降解起到主导作用的活性物种为${{\rm{SO}}_4^ - \cdot } $ ;在光活化体系(hv-Na2S2O8)中,对染料降解起到主导作用的活性物种为${{\rm{SO}}_4^ - \cdot } $ 与OH·;而在亚铁离子活化体系(Fe2+-Na2S2O8)中,OH·对染料降解起主导作用。本文以热活化过硫酸盐体系为代表,探索了亚甲基蓝和甲基橙的降解中间体及路径。在3种活化过硫酸盐体系中,4种阴离子在不同程度上对两种染料的降解均存在抑制作用,尤其在亚铁离子活化过硫酸盐体系中,对4种阴离子的浓度变化更为敏感。Abstract: Advanced Oxidation Processes (AOPs) are widely used in different wastewater treatment. At present, most of these technologies are mainly generated according to hydroxyl radicals. In recent years, sulfate anion (${\rm{SO}}_4^ - \cdot $ ) based AOPs have attracted much attention due to their high reactivity to organic pollutants and high selectivity to complex environmental substrates. But the systematic comparison of various activation modes, as well as the degradation pathways and the effects of different anions on the system were lack of research. In this research, three kinds of activated persulfate systems were systematically compared through the degradation properties of the two dyes, system mechanism and degradation pathway combining radical capture experiments (methanol, tert butyl alcohol) and product analysis, and effect of four different anions (${\rm{HCO}}_3^ - $ , Cl−,${\rm{NO}}_3^ - $ , Br−) on three different persulfates activated systems, were carried out. The results show that${\rm{SO}}_4^ - \cdot $ and OH· have different contributions in different systems, and${\rm{SO}}_4^ - \cdot $ plays a leading role in the heat activation system. Both${\rm{SO}}_4^ - \cdot $ and OH· play a comparable role in the hv activation system, while OH· plays a leading role in the ferrous ion activation system. Here, the heat-activated persulfate system is taken as a representative to explore the degradation intermediates and degradation pathways of methylene blue and methyl orange. Four anions can inhibit the degradation of the two dyes in the three activated persulfate systems, especially in the ferrous ion activated persulfate system, it is more sensitive to the changes of the concentration of four anions.-
Key words:
- persulfate /
- free radical /
- intermediate product /
- anion
-

-
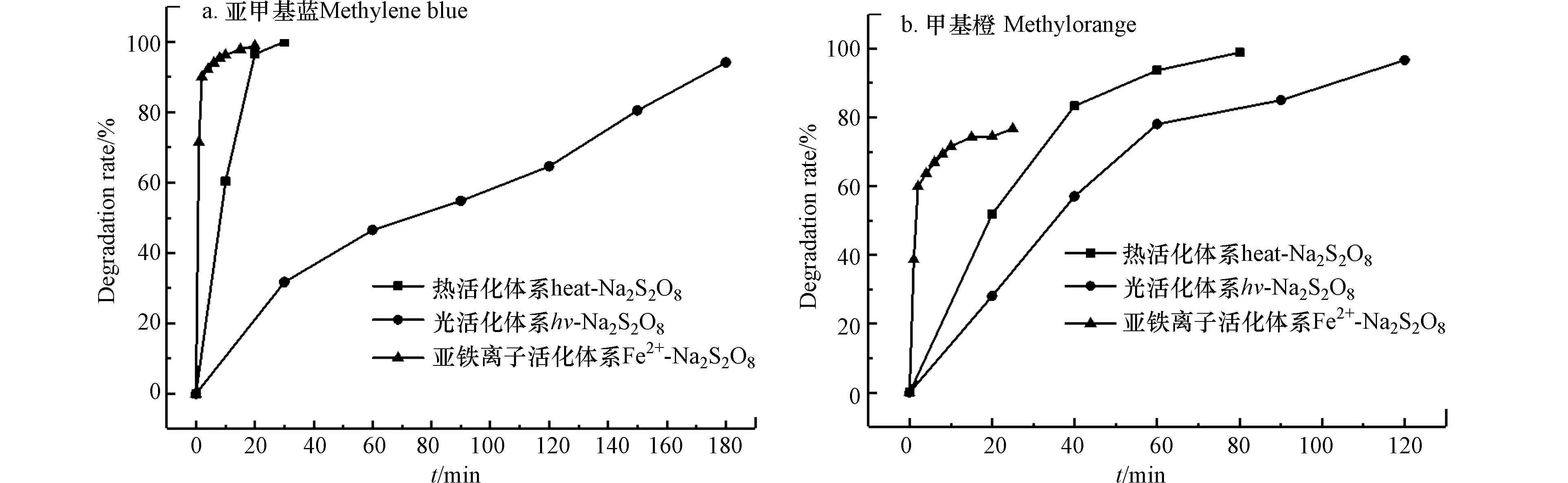
图 1 3种活化过硫酸盐体系中亚甲基蓝和甲基橙的降解
Figure 1. Degradation of methylene blue and methyl orange in three activated persulfate systems
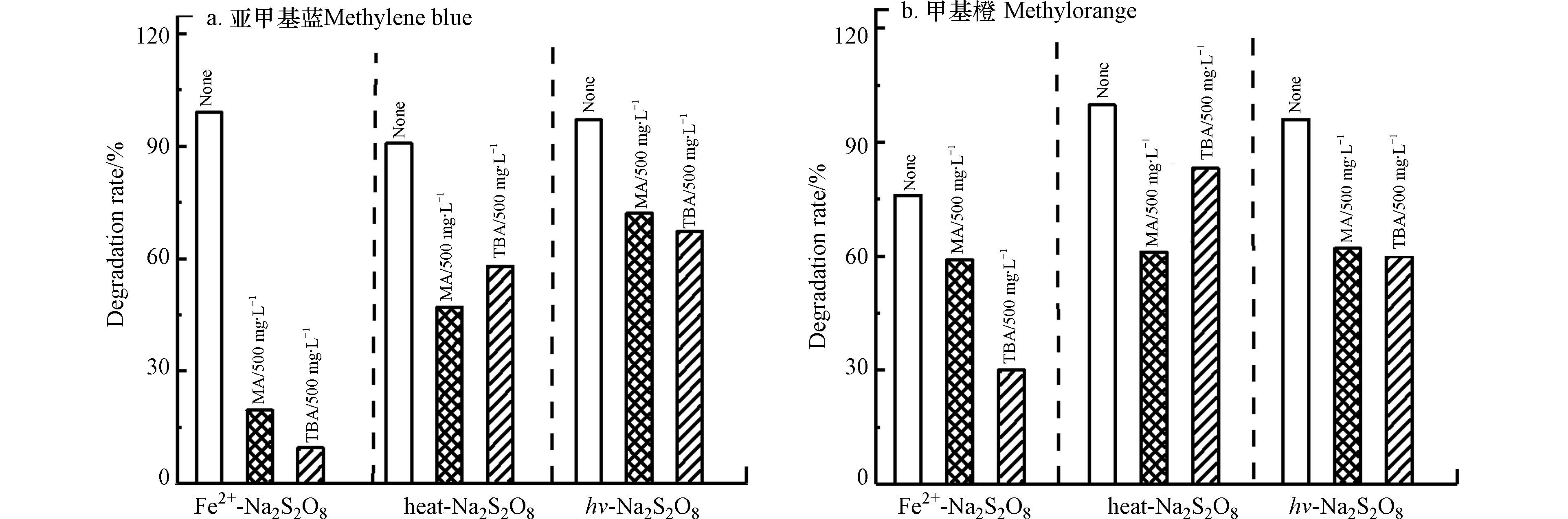
图 2 叔丁醇和甲醇对3种活化过硫酸盐体系中两种染料降解的影响
Figure 2. Effects of tert butyl alcohol and methanol on degradation of two dyes in three activated persulfate systems
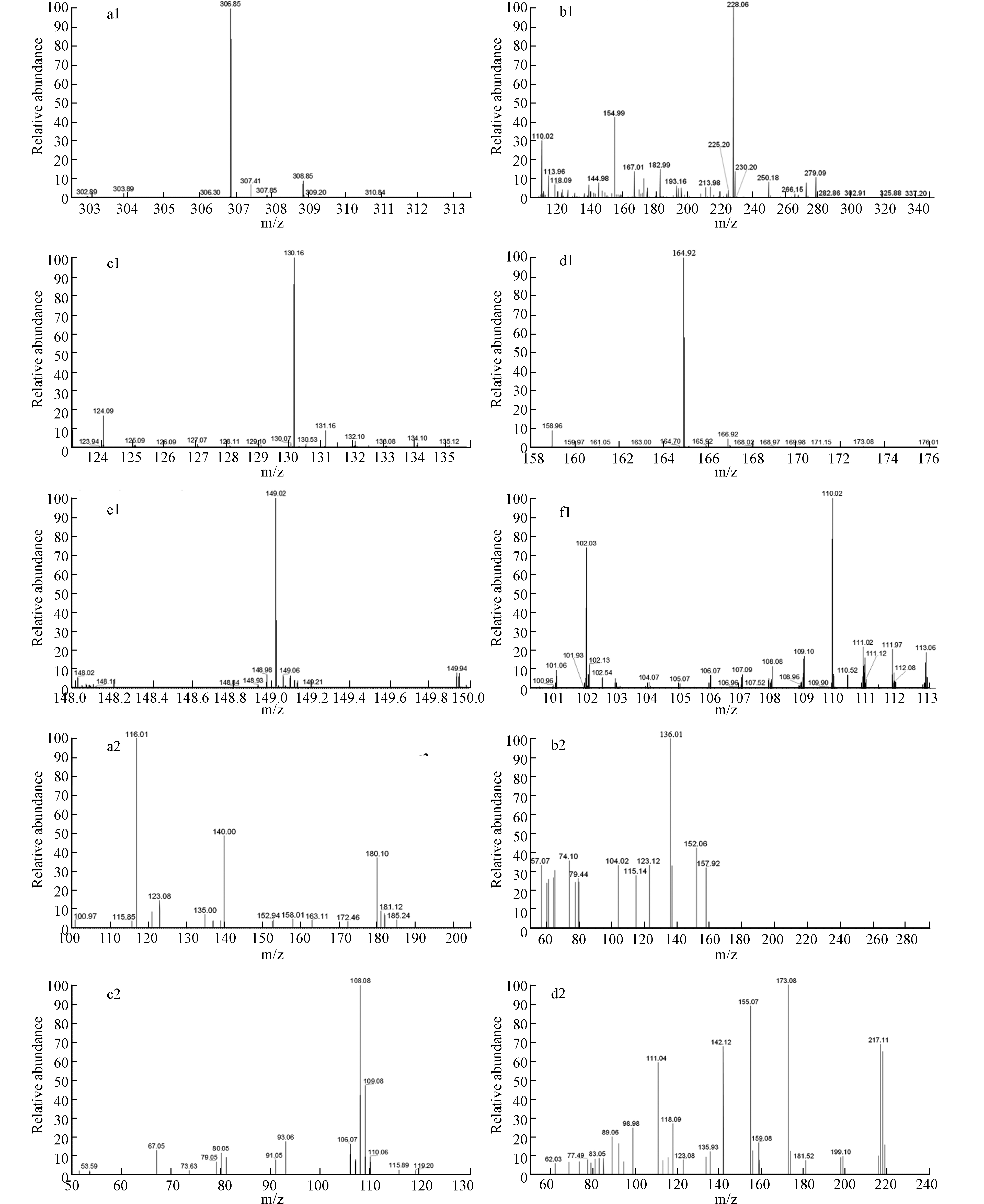
图 3 热活化体系中亚甲基蓝(a1—f1)与甲基橙(a2—d2)中间产物的离子碎片质谱图
Figure 3. Ion fragment mass spectra of methylene blue (a1—f1) and methyl orange (a2—d2) intermediates in heat-activated system

图 4 亚甲基蓝在热活化体系中的降解路径
Figure 4. Degradation pathway of methylene blue in heat-activated system
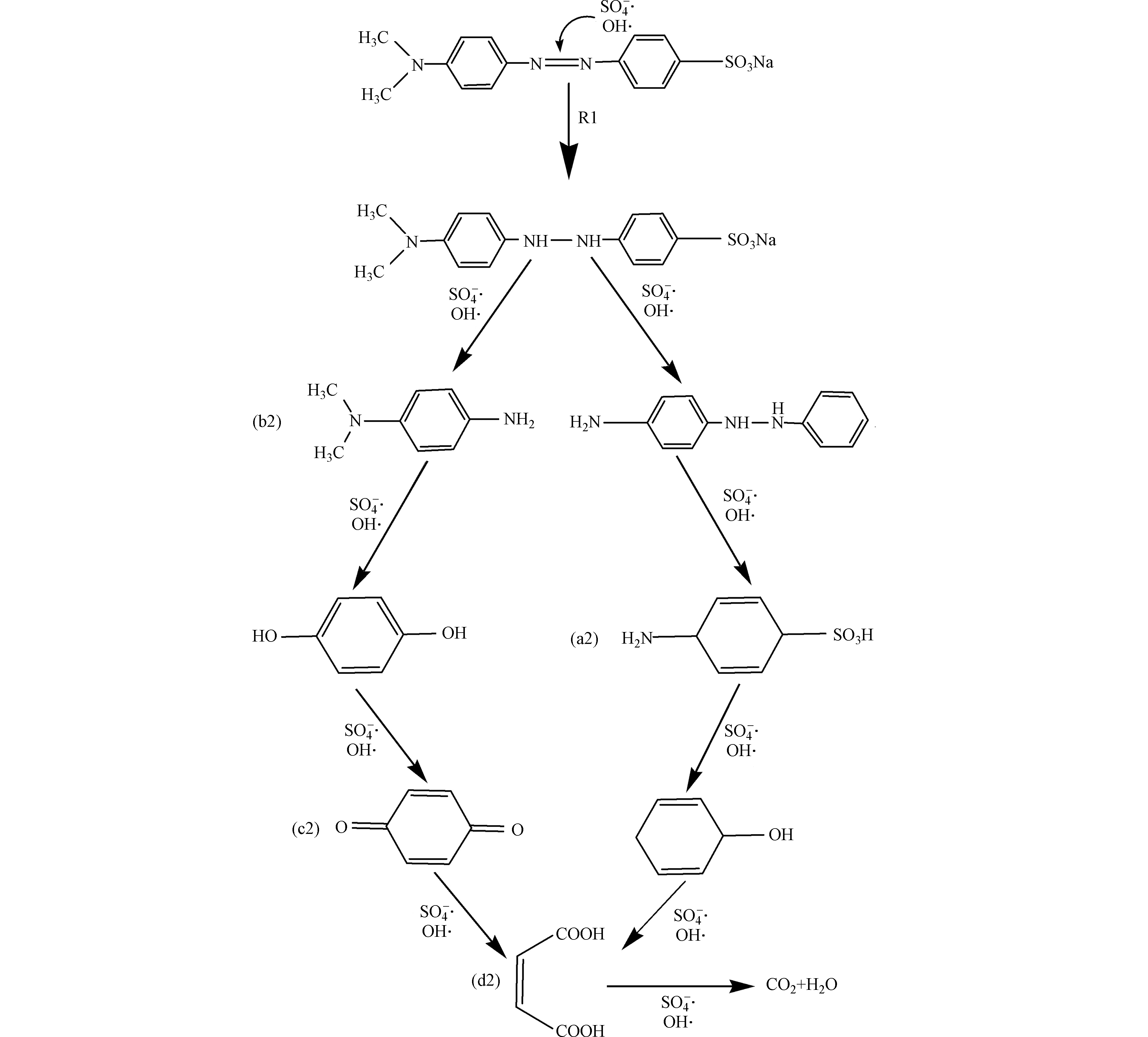
图 6 甲基橙在热活化体系中的降解路径
Figure 6. Degradation pathway of methyl orange in heat-activated system

图 7 3个体系中四种阴离子(
$ {\rm{HCO}}_3^ - $ 、${\rm{NO}}_3^ - $ 、Cl−、Br−)对亚甲基蓝染料的降解影响Figure 7. Effects of four anions (
${\rm{HCO}}_3^ - $ ,${\rm{NO}}_3^ - $ ,Cl−,Br−) on the degradation of methylene blue dye in the three systems表 1 亚甲基蓝降解中间体分析
Table 1. Analysis of degradation intermediates of methylene blue
编号 Number 分子式 Molecular formula 荷质比 Charge-mass ratio 可能结构式 Possible structural formula a1 C16H23N3SO 306.85 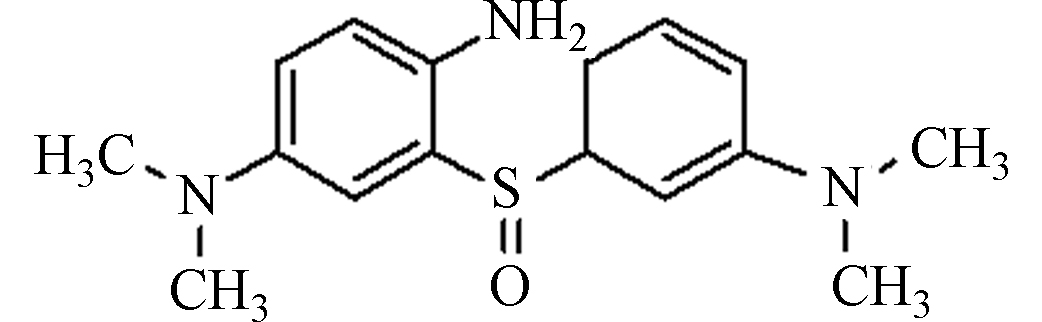
b1 C12H10N3S 228.06 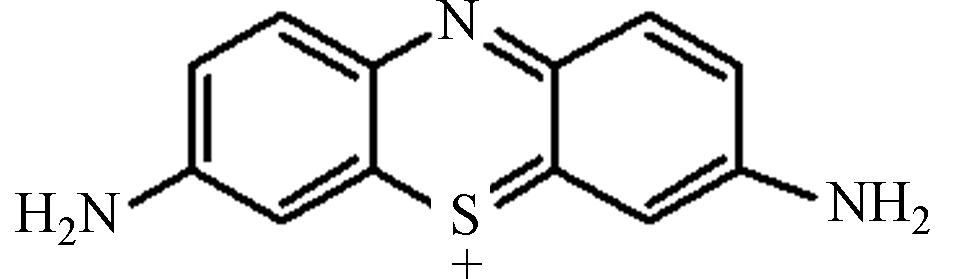
c1 C8H11NSO3Na 224.13 
d1 C8H8N2O2 164.92 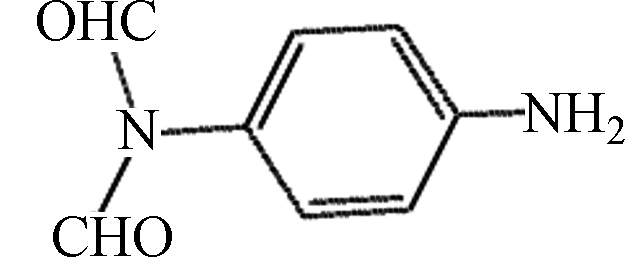
e1 C6H6O3Na 149.02 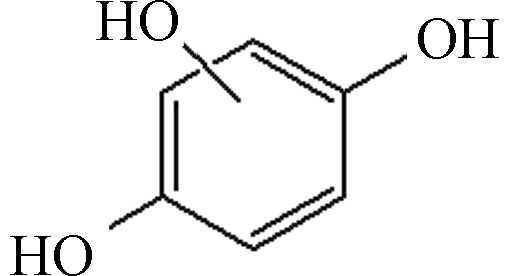
f1 C2H5NO4Na 130.16 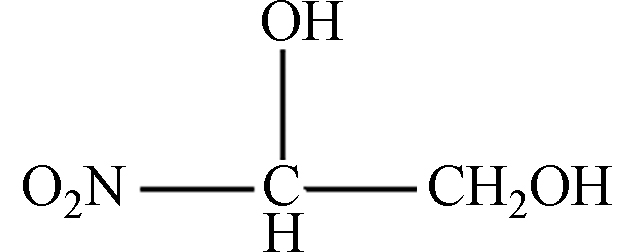
g1 C6H6O2 110.04 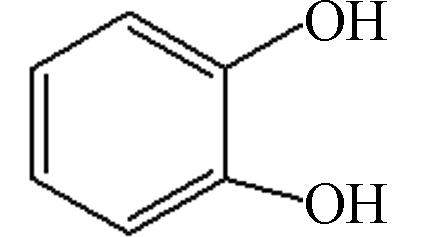
 下载: 导出CSV
下载: 导出CSV
表 2 亚甲基蓝分子轨道活性位点计算
Table 2. Calculation of molecular orbital active sites of methylene blue
EHOMO2 ELUMO2 EHOMO2 + ELUMO2 1C 0.000303352 0.015808328 0.01611168 2C 0.001100568 0.036814232 0.0379148 3C 0.000738513 0.000222207 0.00096072 4C 0.002957525 0.044083627 0.047041152 5C 0.000360553 0.00903695 0.009397503 6C 0.000943767 0.039012747 0.039956514 7N 0.004155314 0.148947152 0.153102466 8C 0.000062021 0.020068239 0.02013026 9C 0.002499575 0.104356398 0.106855973 10C 0.005845132 0.043491797 0.049336929 11C 0.000821844 0.001002979 0.001824822 12C 0.000252401 0.042483703 0.042736104 13C 7.97419E-05 0.026383882 0.026463624 14C 0.000203077 0.046001679 0.046204756 15N 0.000055604 0.052026219 0.052081823 16N 0.001817884 0.032624839 0.034442723 17C 2.85123E-05 0.002840262 0.002868775 18C 0.002126915 0.000551021 0.002677935 19C 4.72912E-05 0.001405402 0.001452693 20C 0.000042666 0.002398119 0.002440785 21Cl 0.547444603 0.01545863 0.562903233
下载: 导出CSV
表 3 甲基橙降解中间体分析
Table 3. Analysis of degradation intermediates of methyl orange
编号 Number 分子式 Molecular formula 荷质比 Charge-mass ratio 可能结构式 Possible structural formula a2 C6H7NO3S 173.08 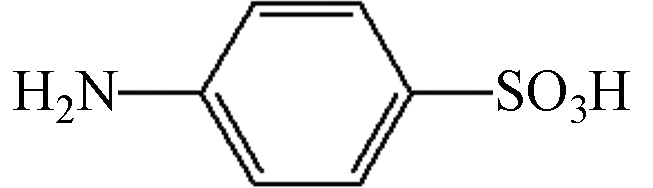
b2 C8H12N2 136.10 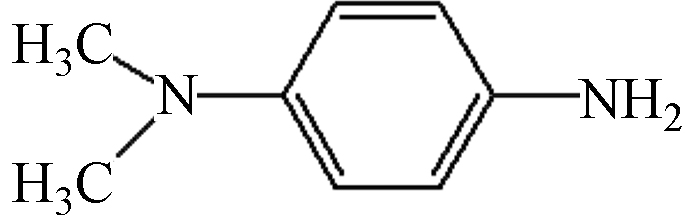
c2 C6H4O6 108.02 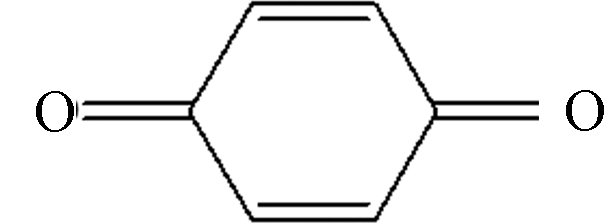
d2 C4H4O4 116.01 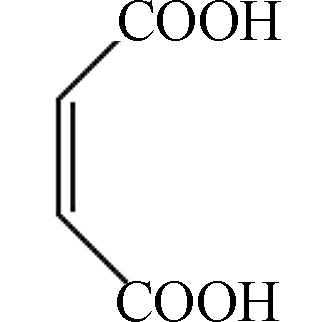 下载: 导出CSV
下载: 导出CSV
表 4 甲基橙分子轨道活性位点计算
Table 4. Calculation of molecular orbital active sites of methyl orange
EHOMO2 ELUMO2 EHOMO2+ ELUMO2 1C 0.0000007808 0.010266 0.0102672538 2C 0.0000035298 0.033624 0.0336275298 3C 0.0000010121 0.034032 0.0340330121 4C 0.0000037059 0.079707 0.0797109959 5C 0.0000011226 0.003621 0.0036218426 6C 0.0000038181 0.070643 0.0706467901 7N 0.0000131172 0.149253 0.1492665612 8N 0.0000018178 0.181302 0.1813035658 9C 0.0001228360 0.00978 0.0099031640 10C 0.0000759619 0.042309 0.0423849619 11C 0.0003032900 0.002045 0.0023486730 12C 0.0009187490 0.052052 0.0529704440 13C 0.0003733960 0.000724 0.0010970590 14C 0.0001624440 0.049068 0.0492307460 15N 0.0000043273 0.022915 0.0229191723 16C 0.0000002569 0.00628 0.0062799189 17C 0.0000002794 0.007449 0.0074492794 18S 0.0000049821 0.000282 0.0002872851 19O 0.1810614660 0.000929 0.1819904660 20O 0.1867748430 0.005771 0.1925458430 21O 0.1868060540 0.005827 0.1926333840
下载: 导出CSV
-
[1] DENG Y, ZHAO R Z. Advanced oxidation processes (AOPs) in wastewater treatment [J]. Current Pollution Reports, 2015, 1(3): 167-176. doi: 10.1007/s40726-015-0015-z [2] MIKLOS D B, REMY C, JEKEL M, et al. Evaluation of advanced oxidation processes for water and wastewater treatment - A critical review [J]. Water Research, 2018, 139: 118-131. doi: 10.1016/j.watres.2018.03.042 [3] CHENG M, ZENG G, HUANG D, et al. Hydroxyl radicals based advanced oxidation processes (AOPs) for remediation of soils contaminated with organic compounds: A review [J]. Chemical Engineering Journal, 2016, 284: 582-598. doi: 10.1016/j.cej.2015.09.001 [4] ZHANG Y, HAN C, ZHANG G, et al. PEG-assisted synthesis of crystal TiO2 nanowires with high specific surface area for enhanced photocatalytic degradation of atrazine [J]. Chemical Engineering Journal, 2015, 268: 170-179. doi: 10.1016/j.cej.2015.01.006 [5] ALI F, KHAN J A, SHAH N S, et al. Carbamazepine degradation by UV and UV-assisted AOPs: Kinetics, mechanism and toxicity investigations [J]. Process Safety and Environmental Protection, 2018, 117: 307-314. doi: 10.1016/j.psep.2018.05.004 [6] QI C D, LIU X T, LI Y, et al. Enhanced degradation of organic contaminants in water by peroxydisulfate coupled with bisulfite [J]. Journal of Hazardous Materials, 2017, 328: 98-107. doi: 10.1016/j.jhazmat.2017.01.010 [7] YANG Q, MA Y, CHEN F, et al. Recent advances in photo-activated sulfate radical-advanced oxidation process (SR-AOP) for refractory organic pollutants removal in water [J]. Chemical Engineering Journal, 2019, 378: 122149. doi: 10.1016/j.cej.2019.122149 [8] GHANBARI F, MORADI M, GOHARI F. Degradation of 2,4, 6-trichlorophenol in aqueous solutions using peroxymonosulfate/activated carbon/UV process via sulfate and hydroxyl radicals [J]. Journal of Water Process Engineering, 2016, 9: 22-28. doi: 10.1016/j.jwpe.2015.11.011 [9] XIAO R, LUO Z, WEI Z, et al. Activation of peroxymonosulfate/persulfate by nanomaterials for sulfate radical-based advanced oxidation technologies [J]. Current Opinion in Chemical Engineering, 2018, 19: 51-58. doi: 10.1016/j.coche.2017.12.005 [10] DEVI P, DAS U, DALAI A K. In-situ chemical oxidation: Principle and applications of peroxide and persulfate treatments in wastewater systems [J]. Science of The Total Environment, 2016, 571: 643-657. doi: 10.1016/j.scitotenv.2016.07.032 [11] DEWIL R, MANTZAVINOS D, POULIOS I, et al. New perspectives for advanced oxidation processes[J]. Journal of Environmental Management, 2017, 195(Pt 2): 93-99. [12] OH W D, DONG Z, LIM T T. Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: Current development, challenges and prospects [J]. Applied Catalysis B:Environmental, 2016, 194: 169-201. doi: 10.1016/j.apcatb.2016.04.003 [13] JI Y F, FAN Y, LIU K, et al. Thermo activated persulfate oxidation of antibiotic sulfamethoxazole and structurally related compounds [J]. Water Research, 2015, 87: 1-9. doi: 10.1016/j.watres.2015.09.005 [14] FERKOUS H, MEROUANI S, HAMDAOUI O, et al. Persulfate-enhanced sonochemical degradation of naphthol blue black in water: Evidence of sulfate radical formation [J]. Ultrasonics Sonochemistry, 2017, 34: 580-587. doi: 10.1016/j.ultsonch.2016.06.027 [15] WANG Q, SHAO Y, GAO N, et al. Degradation kinetics and mechanism of 2, 4-Di-tert-butylphenol with UV/persulfate [J]. Chemical Engineering Journal, 2016, 304: 201-208. doi: 10.1016/j.cej.2016.06.092 [16] CHENG X X, WU D J, LIANG H, et al. Effect of sulfate radical-based oxidation pretreatments for mitigating ceramic UF membrane fouling caused by algal extracellular organic matter [J]. Water Research, 2018, 145: 39-49. doi: 10.1016/j.watres.2018.08.018 [17] LIU Y, WANG S, WU Y, et al. Degradation of ibuprofen by thermally activated persulfate in soil systems [J]. Chemical Engineering Journal, 2019, 356: 799-810. doi: 10.1016/j.cej.2018.09.002 [18] MATZEK L W, CARTER K E. Activated persulfate for organic chemical degradation: A review [J]. Chemosphere, 2016, 151: 178-188. doi: 10.1016/j.chemosphere.2016.02.055 [19] SHAH N S, ALI KHAN J, SAYED M, et al. Hydroxyl and sulfate radical mediated degradation of ciprofloxacin using nano zerovalent manganese catalyzed S2O82− [J]. Chemical Engineering Journal, 2019, 356: 199-209. doi: 10.1016/j.cej.2018.09.009 [20] LIN C C, TSAI C W. Degradation of isopropyl alcohol using UV and persulfate in a large reactor [J]. Separation and Purification Technology, 2019, 209: 88-93. doi: 10.1016/j.seppur.2018.06.068 [21] JIANG X X, WU Y L, WANG P, et al. Degradation of bisphenol A in aqueous solution by persulfate activated with ferrous ion [J]. Environmental Science and Pollution Research, 2013, 20(7): 4947-4953. doi: 10.1007/s11356-013-1468-5 [22] KANG S, QIN L, ZHAO Y, et al. Enhanced removal of methyl orange on exfoliated montmorillonite/ chitosan gel in presence of methylene blue [J]. Chemosphere, 2020, 238(Jana): 124693.1-124693.7. [23] ZHU S S, LI X J, KANG J, et al. Persulfate activation on crystallographic manganese oxides: Mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants [J]. Environmental Science & Technology, 2019, 53(1): 307-315. [24] QI C D, LIU X T, MA J, et al. Activation of peroxymonosulfate by base: Implications for the degradation of organic pollutants [J]. Chemosphere, 2016, 151: 280-288. doi: 10.1016/j.chemosphere.2016.02.089 [25] ZHOU Z, LIU X, SUN K, et al. Persulfate-based advanced oxidation processes (AOPs) for organic-contaminated soil remediation: A review [J]. Chemical Engineering Journal, 2019, 372: 836-851. doi: 10.1016/j.cej.2019.04.213 [26] MILH H, SCHOENAERS B, STESMANS A, et al. Degradation of sulfamethoxazole by heat-activated persulfate oxidation: Elucidation of the degradation mechanism and influence of process parameters [J]. Chemical Engineering Journal, 2020, 379: 122234. doi: 10.1016/j.cej.2019.122234 [27] DULOVA N, KATTEL E, TRAPIDO M. Degradation of naproxen by ferrous ion-activated hydrogen peroxide, persulfate and combined hydrogen peroxide/persulfate processes: The effect of citric acid addition [J]. Chemical Engineering Journal, 2017, 318: 254-263. doi: 10.1016/j.cej.2016.07.006 [28] DA X, JI H, ZHAO Z, et al. Strongly prolonged hydroxyl radical production for Fenton-like reactions: The golden touch of Cu [J]. Separation and Purification Technology, 2019, 213: 500-506. doi: 10.1016/j.seppur.2018.12.060 [29] LI M N, ZHANG Z H, WU S Y, et al. DFT calculations of the surface and the electronic structure of silicon core approximants for sensing [J]. Optik, 2017, 148: 344-349. doi: 10.1016/j.ijleo.2017.09.022 [30] 胡嘉敏, 张静, 袁琳, 等. 紫外强化铜循环催化过硫酸盐降解甲基橙 [J]. 环境科学研究, 2018, 31(1): 123-129. [31] CHOU Y C, LO S L, KUO J, et al. Microwave-enhanced persulfate oxidation to treat mature landfill leachate [J]. Journal of Hazardous Materials, 2015, 284: 83-91. doi: 10.1016/j.jhazmat.2014.10.043 [32] JIANG M D, LU J H, JI Y F, et al. Bicarbonate-activated persulfate oxidation of acetaminophen [J]. Water Research, 2017, 116: 324-331. doi: 10.1016/j.watres.2017.03.043 [33] GUO Y, ZENG Z, LI Y, et al. In-situ sulfur-doped carbon as a metal-free catalyst for persulfate activated oxidation of aqueous organics [J]. Catalysis Today, 2018, 307: 12-19. doi: 10.1016/j.cattod.2017.05.080 [34] LI J, REN Y, JI F, et al. Heterogeneous catalytic oxidation for the degradation of p-nitrophenol in aqueous solution by persulfate activated with CuFe2O4 magnetic nano-particles [J]. Chemical Engineering Journal, 2017, 324: 63-73. doi: 10.1016/j.cej.2017.04.104 [35] MA J, YANG Y Q, JIANG X, et al. Impacts of inorganic anions and natural organic matter on thermally activated persulfate oxidation of BTEX in water [J]. Chemosphere, 2018, 190: 296-306. doi: 10.1016/j.chemosphere.2017.09.148 [36] ROY K, AGARKOTI C, MALANI R S, et al. Mechanistic study of sulfadiazine degradation by ultrasound-assisted Fenton-persulfate system using yolk-shell Fe3O4@hollow@mSiO2 nanoparticles [J]. Chemical Engineering Science, 2020, 217: 115522. doi: 10.1016/j.ces.2020.115522 [37] CHEN Y Q, DENG P Y, XIE P C, et al. Heat-activated persulfate oxidation of methyl- and ethyl-parabens: Effect, kinetics, and mechanism [J]. Chemosphere, 2017, 168: 1628-1636. doi: 10.1016/j.chemosphere.2016.11.143 [38] DU X, ZHANG Y, SI F, et al. Persulfate non-radical activation by nano-CuO for efficient removal of chlorinated organic compounds: Reduced graphene oxide-assisted and CuO (001) facet-dependent [J]. Chemical Engineering Journal, 2019, 356: 178-189. doi: 10.1016/j.cej.2018.08.216 [39] ZHANG W Q, ZHOU S Q, SUN J L, et al. Impact of chloride ions on UV/H2O2 and UV/persulfate advanced oxidation processes [J]. Environmental Science & Technology, 2018, 52(13): 7380-7389. [40] ZHOU L, FERRONATO C, CHOVELON J M, et al. Investigations of diatrizoate degradation by photo-activated persulfate [J]. Chemical Engineering Journal, 2017, 311: 28-36. doi: 10.1016/j.cej.2016.11.066 [41] WANG L, JI Y F, LU J H, et al. Comparative study of the formation of brominated disinfection byproducts in UV/persulfate and UV/H2O2 oxidation processes in the presence of bromide [J]. Environmental Science and Pollution Research International, 2017, 24(29): 23219-23225. doi: 10.1007/s11356-017-9935-z [42] LU J, WU J, JI Y, et al. Transformation of bromide in thermo activated persulfate oxidation processes [J]. Water Research, 2015, 78: 1-8. doi: 10.1016/j.watres.2015.03.028 [43] SONG Q, FENG Y, LIU G, et al. Degradation of the flame retardant triphenyl phosphate by ferrous ion-activated hydrogen peroxide and persulfate: Kinetics, pathways, and mechanisms [J]. Chemical Engineering Journal, 2019(361): 929-936. -
 点击查看大图
点击查看大图
计量
- 文章访问数: 5824
- HTML全文浏览数: 5824
- PDF下载数: 166
- 施引文献: 0










































































































































