






























































































-
大气固态水,包括冰晶和雪颗粒在内,在大气对流层以及高纬度和高海拔地区的寒冷生态系统中广泛存在[1-2]. 冰/雪具有较大的比表面积使其有着很强的吸附能力[3-5]. 冰/雪可以吸附大气污染物,再通过物理或化学相互作用[6-8]改变它们的电子结构特征,进而影响其迁移和转化,最终影响大气污染物的环境归趋[9-12]. 因此,了解大气污染物在冰表面的吸附行为对其环境风险评估具有重要意义.
近年来,大气中甲醇、草酸和甲酰胺等低分子量小分子有机污染物在冰表面的吸附行为已被广泛研究,然而这些小分子化合物的大气寿命较短,持久性较低,与大气中冰表面发生相互作用的机会较小[13-16]. 相对于这些低分子量的有机化合物,分子高持久性强的有机污染物,如芳香类化合物,更有机会被吸附在冰表面进而影响其大气迁移、转化行为. 芳香类化合物作为一类重要的持久性有机污染物,其种类多,来源广,在大气环境中具有较高的浓度水平[17-20]. 芳香类化合物在大气冰表面的吸附转化行为会显著影响其环境归趋和潜在的环境风险. 目前关于芳香类化合物,如苯、苯甲醛、苯胺和硝基苯在冰表面吸附行为的研究已有开展[21-24],结果表明不同的取代基通过改变芳香类化合物与冰表面的作用方式,包括静电相互作用、氢键相互作用类型和强度等,进而影响其在冰表面的吸附量,吸附能和吸附等温线等,最终影响芳香类化合物在冰表面的吸附行为. 因此,为了进一步理解芳香类化合物在冰表面的吸附行为,更多的研究应该关注其他具有不同取代基团的芳香类化合物在冰表面上的吸附行为.
苯酚是一种含羟基(—OH)的有毒芳香类化合物. 苯酚作为重要的化工原料,在石油行业、焦炭冶金工业和生产酚醛树脂等方面应用广泛,而且会在其生产、使用和运输过程中不可避免地排放到环境中[25]. 此外,生物质燃烧以及汽车尾气排放也是大气中苯酚的重要来源[26-27]. 苯酚在城市空气中的平均浓度约为0.13 μg·cm−3[28]. 同时,苯酚已被我国生态环境部列为优先控制污染物,被美国环境保护署列为有害空气污染物. 苯酚进入大气环境后,发生光化学降解是其在大气环境中重要的去除途径. 最近的研究发现苯酚在冰表面比在溶液中具有更快的光降解速率. 一种可能的解释是,当苯酚分子在气-冰界面被吸附时,它们的紫外可见吸收光谱发生了红移,暗示了苯酚在大气冰表面的吸附可能会改变其大气转化行为[29]. 此外,分子动力学模拟温度条件为263 K时单个苯酚分子在冰表面的吸附行为也发现,苯酚在气-水界面的吸附比溶解在水液滴内部热力学更可行. 然而目前对于苯酚在低温条件下冰表面的吸附行为还不清楚.
本文使用巨正则蒙特卡罗模拟(Grand Canonical Monte Carlo (GCMC))来研究苯酚在冰表面的吸附行为[14-15,21-24,30]. 选择可靠的力场是GCMC模拟成功的关键,冰的分子力场使用可以很好地描述固态水理化性质的TIP5P水模型[31-33],苯酚的分子力场使用基于OPLS-AA的优化力场[34-36]. 基于模拟结果,计算和分析了苯酚在冰表面的吸附等温线,并从苯酚在冰表面的吸附方向、氢键相互作用和吸附能等方面探讨了不同吸附阶段的苯酚的吸附特征. 本研究结果对理解苯酚在冰表面的吸附行为以及对其他含—OH的芳香类化合物在冰表面的吸附行为具有重要意义.
-
类似于本课题组关于硝基苯和苯胺在冰表面吸附的研究[23-24],使用“改变原子电荷并保持 Lennard-Jones(L-J)势能参数不变”的方法[37]来确定最佳的苯酚分子力场. 苯酚的L-J参数来自OPLS-AA力场,选择苯酚原子电荷的标准是将苯酚的原子电荷乘以倍数因子进行调整,配合TIP5P水分子力场进行分子动力学模拟,寻找可以很好地模拟苯酚水溶解自由能实验值的苯酚原子电荷. 通过量子化学计算,利用约束静电势(RESP)方法拟合HF/6-31G*水平下计算的静电势,得到苯酚力场参数优化所使用的初始原子电荷[37].
通过平均力势能(potential of mean force, PMF)计算获得苯酚的水溶解自由能(ΔG*)[38]. PMF 计算设置的细节类似于本课题组对硝基苯和苯胺在冰表面吸附的研究[23-24]. 用于自由能计算的模拟盒子如图1(A)所示,该模拟盒子的长宽高分别为3.09、3.09、20 nm,与前人进行力场优化采用的盒子参数一致[23-24] ,盒子内包含预先平衡的970个水分子,距离水表面2.5 nm 处放置的定位原子,并在距离水表面1.5 nm 处放置苯酚分子. 使用伞型采样方法的分子动力学模拟计算从气相移动单个苯酚分子通过水相再回到气相的自由能变化. 本研究中,分子动力学模拟使用 GROMACS 4.5.5 程序进行[39].
-
本研究以六边形冰(Ih)的0001表面作为冰表面模型[40-41],选择具有5个点位的TIP5P分子力场[31-33]模拟冰相中的水分子,苯酚分子力场采用1.1节中优化的力场. Ih冰是大气对流层最常见的冰晶类型[41-43],Ih冰的0001表面是目前用于研究大气污染物在冰表面的吸附行为使用最普遍的冰表面[22-24]. 基于大气中对流层上层(温度在188—233 K之间)冰(卷云)和冰/水混合云所处的真实环境条件[22],本研究采用200 K作为模拟体系的温度,该温度条件已被成功用于多种大气污染物在冰表面吸附行为的理论和实验研究[13,16,21,44]. 值得注意的是,在温度高于200 K直至近零点的271 K条件下,冰表面会出现无序的似液体层[45-47]. 因此,在200 K温度条件下的GCMC模拟结果不能外推更高温度条件下苯酚在冰表面的吸附行为.
根据研究体系大小建立了沿X、Y、Z分别为10、3.593、3.889 nm的GCMC模拟盒子,与前人模拟芳香类化合物在冰表面吸附行为的模拟盒子设置一致[22-24]. 模拟盒子中的冰相由包含2880个水分子的18个水分子层沿X轴排列在具有三维周期性边界条件的模拟盒子中间. 为获得苯酚的吸附等温线,进行了46次独立的GCMC模拟. 模拟中,苯酚的化学势(μ)变化范围为−72.88 kJ·mol−1至−56.92 kJ·mol−1,涵盖了从冰表面没有苯酚分子吸附到模拟盒子被苯酚分子填满的吸附情况.
本研究使用了Mezei开发的(Metropolis Monte Carlo Program, MMC program)程序进行GCMC模拟[32,48]. 体系的总势能是所有分子对的相互作用能的总和,设置相互作用截止距离为12.50 Å. 在模拟过程的插入/删除步骤中,使用Mezei开发的空腔有偏算法[49]确定体系中苯酚分子的插入或删除. 模拟过程中,苯酚分子吸附在冰表面的位移和插入/删除动作交替进行. 在随机选择的分子位移步骤中,分子的随机位移不超过0.025 nm,并且在一个随机选择的空间中围绕固定轴随机旋转,旋转角度不超过15°. 苯酚分子吸附在冰表面,沿100×100×100的网格域搜索空腔,每经过1×106蒙特卡罗步后重新生成空腔. 模拟共运行了6×108步,4×108步用于体系平衡,2×108步用于吸附过程,每5×105步模拟后选取代表性构型进行后续分析.
-
采用48-H2O冰模型作为量子化学计算的冰表面模型[23-24]. 该冰模型由Ih冰的0001表面切割的48个水分子组成,分两层排布,与前人研究所使用的冰模型一致[23-24,41]. 在M062X/6-31+G(d,p)水平下进行几何优化和频率计算,基于优化水平下的构型,在M062X/6-311++G(3df,2pd)水平下进行单点能量计算. 通过对比不同的吸附构型的吉布斯自由能,选取吉布斯自由能最低的结构作为最稳定的吸附构型. 量子化学计算在Gaussian 09程序包[50]中进行.
-
使用多种苯酚分子力场,计算得到苯酚穿过TIP5P水盒子的PMF曲线如图图1(B)所示. 其中,“PMF 1”代表由苯酚OPLS-AA的L-J参数与RESP电荷组成的力场计算得到的自由能分布曲线; “PMF 2”代表由苯酚OPLS-AA的L-J参数与苯酚RESP电荷×倍数因子1.15组成的力场计算得到的自由能分布曲线; “PMF 3”代表由苯酚OPLS-AA的L-J参数与苯酚RESP电荷×倍数因子1.20组成的力场计算得到的自由能分布曲线. 不同力场参数下的自由能曲线差别较大,但所有自由能曲线都具有一般特性,即气-水界面处的自由能低于水相中的自由能,表明苯酚优先吸附在气-水界面. 苯酚在液态水内与气相之间自由能的差值代表了水溶解自由能值(ΔG*). 在298 K下,苯酚的水溶解自由能ΔG*的实验值取3个实验获得的亨利定律常数计算值的平均值[51],见图1(B)中的蓝色水平虚线. 对比发现,PMF2预测的ΔG*值与实验值吻合最好,而其他的苯酚力场高估或低估了ΔG*值. 因此,选择苯酚OPLS-AA力场的L-J势能参数和RESP电荷×1.15的电荷参数来作为苯酚的优化力场,详见表1.
为了进一步验证苯酚优化力场的合理性,基于优化力场计算了苯酚的偶极矩、密度、蒸发焓和热容,详见表2. 相比于OPLS-AA原始力场,基于优化力场计算的偶极矩、蒸发焓和热容与实验值[35,52-54]更吻合. 尽管原始OPLS-AA力场计算的密度更接近实验密度值,但与优化力场计算的密度值差异非常小. 鉴于计算值与实验值在密度、蒸发焓和热容方面的良好一致性,表明优化力场可以很好地描述苯酚的分子间相互作用,且与实验值一致的水溶解自由能值可以很好地描述苯酚和TIP5P水分子之间的相互作用. 因此,结合苯酚优化的分子力场和TIP5P水模型可以很好模拟苯酚在冰表面的吸附行为.
-
苯酚在冰表面吸附的平均个数
$\left\langle {{N}} \right\rangle$ 与化学势μ之间的关系通过吸附等温线表示,呈现在图2(A),详细数据在表3中列出. 对应的吸附构型快照,包括俯视图和侧视图均呈现于图3. 如图2(A)所示,苯酚分子在冰表面的吸附量随化学势的增长而增加.化学势从−72.88 kJ·mol−1至−71.55 kJ·mol−1时,苯酚在冰表面的吸附数量缓慢增加,直至化学势达到−71.55 kJ·mol−1(PhOH Ⅰ)时,苯酚分子吸附量明显增加. 当化学势达到−69.89 kJ·mol−1(PhOH Ⅱ)时,苯酚在冰表面吸附量增长趋势变缓. 当化学势从−58.08 kJ·mol−1到−56.59 kJ·mol−1(PhOH Ⅲ到PhOH Ⅳ)时,苯酚在冰表面的吸附量快速增加,随后吸附等温线突跃到一个恒定的常数平台(PhOH Ⅳ),此时模拟盒子中的冰表面上方已经充满苯酚分子. 因此,凝结点处在PhOH Ⅲ和PhOH Ⅳ之间. 苯酚在冰表面的吸附等温线呈现一种“双平台”模式,随着苯酚在冰表面的吸附量增加,出现由接近单层吸附饱和至单层吸附饱和的平台区,再随着苯酚达到吸附凝结点,呈现出凝聚相的平台区. 苯酚的吸附等温线不同于苯和硝基苯的吸附等温线,与苯甲醛和苯胺在冰表面的吸附等温线相似[22-23].
为了进一步研究苯酚在冰表面上的吸附行为,将<N>和μ的函数转化为Γ和Prel的函数. Γ是苯酚在冰表面的密度,Prel是相对压力,由气相压力和凝结点的压力P0归一化得到. 转化后的Γ和Prel型等温线仅在凝结点之前有意义,当Prel大于1之后将失去物理意义. 函数的转化通过下面两个公式实现[13].
式(1)中的YZ 为冰表面的横截面积,系数 2 代表模拟盒子的两个冰表面.
式(2)中kB为玻尔兹曼常数,μ0为吸附凝结点前对应的化学势值,苯酚的μ0值为−57.59 kJ·mol−1.
对图2(B)中表面密度与相对压力的曲线拟合Langmuir吸附等温线,发现吸附等温线在整个考虑的压力范围内都没有很好地表现出Langmuir特性,这意味着它不能完全满足Langmuir吸附的两个先决条件. 条件一要求饱和吸附层为严格的单分子层,条件二要求吸附质之间无相互作用. 当图2(B)插图中所示的Prel低于0.0008时,吸附等温线拟合非常好(R2=0.99),此时被吸附的苯酚分子独立占据冰表面上的吸附位点并与冰表面相互作用. 说明随着苯酚分子吸附在冰表面的数量增加,吸附等温线与Langmuir吸附等温线开始发生偏差.
为了分析苯酚在冰表面的吸附层特征,计算了PhOH Ⅰ—Ⅳ被吸附的苯酚分子中与—OH相连碳原子(ipso碳原子)的个数密度分布,呈现在图4(A). 由图4(A)所示,对于体系PhOH Ⅰ和PhOH Ⅱ,ipso碳原子的分布曲线均为单峰,且两个体系的密度峰位置几乎相同. 然而PhOH Ⅱ峰值比PhOH Ⅰ峰值更高,且密度峰值位置更宽,表明PhOH Ⅱ体系中吸附的苯酚离冰表面更远. 对于PhOH Ⅲ,在主密度峰的外侧出现另一个较低的峰(X≈5.4—5.6 nm),表明苯酚在冰表面单层吸附饱和后开始了多层吸附. 与早期的研究类似,在PhOH Ⅲ和密度分布曲线中,第一个最低谷作为苯酚在冰表面上吸附的第一吸附层(图4(A)中橙色虚线标出)[22-23]. 对于PhOH Ⅳ,主密度峰外侧存在多个较低的峰,表明此时苯酚已经进入凝聚相,属于过冷液体. 在后续探究苯酚的吸附行为中,只分析了PhOH Ⅰ—Ⅲ中苯酚在冰表面的吸附方向和第一吸附层中苯酚与冰表面的相互作用能,对PhOH Ⅳ体系的吸附构型和吸附能等不作分析.
苯酚在冰表面的方向变化分布曲线呈现在图4(B)中. 从图4(B)可以看出,当苯酚在冰表面的吸附量很少时(PhOH Ⅰ),苯酚分子与冰表面主要形成约66°的倾斜角(
$ \mathrm{c}\mathrm{o}\mathrm{s}\gamma \approx 0.4 $ ). 此时苯酚的—OH与冰表面水分子之间存在氢键相互作用,这与已有的芳香类化合物优先平行吸附在冰表面的吸附行为不同[21-24]. 随着冰表面上的苯酚数量逐渐增加至吸附单层饱和后,苯酚分子与冰表面之间形成的角度从0°至90°均有分布,平行吸附于冰面的分子数量增多. 通过观察吸附于冰表面上的苯酚分子构型,发现平行吸附的苯酚分子与冰表面水分子形成了O—H···π键,需要指出的是:平行于冰表面不是苯酚主要的吸附方向,即使在单层吸附饱和的PhOH Ⅲ,平行吸附的苯酚分子数量在总的吸附分子数量中占比不足30%.为揭示苯酚吸附在冰表面的吸附能信息,计算了苯酚与冰表面相互作用能(
$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ )、苯酚分子之间相互作用能($ {U}_{\mathrm{b}}^{\mathrm{P}\mathrm{h}\mathrm{O}\mathrm{H}} $ )和苯酚分子与冰表面的总结合能($ {U}_{\mathrm{b}}={U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}}+{U}_{\mathrm{b}}^{\mathrm{P}\mathrm{h}\mathrm{O}\mathrm{H}} $ ).图5呈现了
$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ 、$ {U}_{\mathrm{b}}^{\mathrm{P}\mathrm{h}\mathrm{O}\mathrm{H}} $ 和$ {U}_{\mathrm{b}} $ 在PhOH Ⅰ—Ⅲ中的分布情况. 从图5可以看出,在PhOH Ⅰ体系中,$ {U}_{\mathrm{b}}^{\mathrm{P}\mathrm{h}\mathrm{O}\mathrm{H}} $ 的分布在0 kcal·mol−1附近有一个高尖峰,说明苯酚分子独立吸附在冰表面,苯酚分子间不存在相互作用. 因此,$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ 主要贡献了PhOH Ⅰ中的$ {U}_{\mathrm{b}} $ ,计算$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ 的值为−18.25 kcal·mol−1. 相比于PhOH Ⅰ,PhOH Ⅱ—Ⅲ中的$ {U}_{\mathrm{b}}^{\mathrm{P}\mathrm{h}\mathrm{O}\mathrm{H}} $ 向着能量更低的方向移动,而$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ 向着能量更高的方向移动. 这表明随着苯酚吸附量的增加,苯酚分子间的相互作用增强,导致吸附分子与冰表面的相互作用减弱. 这种分子之间存在的相互作用是造成拟合曲线偏离Langmuir吸附方程(图2(B))的原因. -
前文对苯酚在冰表面的吸附方向分析发现,苯酚的—OH与冰表面水分子之间存在氢键相互作用,这可能会影响苯酚在冰表面的吸附方向和吸附能. 因此,本部分重点研究了苯酚的—OH与冰表面水分子之间形成的氢键类型和概率分布. 统计发现,苯酚的—OH基团与冰表面水分子之间形成的氢键有3种类型,呈现于图6(A). Type1中苯酚—OH上的H原子与冰表面水分子上的O原子形成氢键(O—H···O);Type2中苯酚—OH上的O原子与冰表面水分子上的H原子形成氢键(O···H—O);Type3中苯酚—OH上的H原子与冰表面水分子上的O原子形成氢键且—OH上的O原子与冰表面水分子上的H原子形成氢键. 该部分使用两个距离标准和一个角度条件定义氢键:氢键受体和供体两个重原子之间的距离在< 0.35 nm(图6(A)蓝色虚线),供体H原子和受体O原子之间的距离< 0.245 nm(图6(A)红色虚线)以及O—H···O/O···H—O角度≥150°[55-57].
上述3种氢键类型的概率分布呈现于图6(B),Type1在PhOH Ⅰ—Ⅲ中始终占据主导地位,即苯酚—OH上的H原子与冰表面水分子上的O原子形成氢键(O—H···O)是最主要的氢键类型. 当吸附量很少(体系 PhOH Ⅰ)时,这种优势效应更加明显,Type1占总吸附数的98.5%,这也与前文苯酚分子在冰表面的方向分析结果相一致. Type2在PhOH Ⅰ中占比为1.5%,在PhOH Ⅱ 和PhOH Ⅲ中的占比明显多于PhOH Ⅰ,分别为7.8%和6.5%. PhOH Ⅰ中不存在Type3,Type3在PhOH Ⅱ 和PhOH Ⅲ中也只少量存在,分别占PhOH Ⅱ和PhOH Ⅲ的0.0048%和0.09%,远小于前两种氢键类型.
-
为进一步确认采用的分子力场的可靠性,本研究采用量子化学计算方法研究了苯酚与冰表面的相互作用能,并与GCMC模拟结果进行对比. 通过对比不同吸附构型的吉布斯自由能,选取吉布斯自由能最低的结构作为最稳定的吸附构型,如图7所示。在M062X/6-31+G(d,p)//M06-2X/6-311++G(3df, 2pd)水平下,计算得到苯酚和冰表面之间的相互作用能为−18.85 kcal·mol−1,与GCMC模拟结果(−18.25 kcal·mol−1)吻合较好,进一步验证了使用的分子力场和GCMC结果的可靠性.
-
本研究进一步探讨了不同取代基对芳香类污染物在冰表面吸附的影响,比较了苯酚与苯、苯胺、硝基苯和苯甲醛在冰表面的吸附行为[21-24]. 对比发现,苯酚的吸附等温线与苯和硝基苯的吸附等温线有很大的不同. 苯和硝基苯的吸附等温线在达到吸附凝结点前,吸附量随相对压力增加而增加. 而苯酚的吸附等温线呈现一种“双平台”模式,与苯甲醛和苯胺在冰表面的吸附等温线较为类似. 对比吸附在冰表面的分子方向发现,苯酚在冰表面的吸附量很少时,苯酚分子与冰表面主要形成约66°的倾斜角,这与苯、苯甲醛、硝基苯和苯胺都优先平行吸附在冰表面不同. 此外,从吸附能来看,吸附量少的条件下,
$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ 的大小顺序为苯酚(−18.25 kcal·mol−1)<硝基苯(−18.16 kcal·mol−1)<苯胺(−15.77 kcal·mol−1)<苯甲醛(−14.10 kcal·mol−1). 通过对比,发现苯酚在冰表面的$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ 是最低的,说明苯酚的—OH与冰表面具有较强的相互作用,也表明不同取代基会对芳香类化合物在冰表面的吸附行为产生重要影响. -
本文应用了巨正则蒙特卡罗模拟,详细分析了温度为200 K条件下苯酚在冰表面的吸附特征. 研究表明,苯酚吸附在冰表面形成的吸附等温线呈现“双平台”模式,相对分压较小时,苯酚吸附个数快速增加,达到第一个单层饱和平台期;达到吸附凝结点后,苯酚填充了整个模拟盒子,吸附个数基本不变,达到第二个平台期. 由于冰表面吸附分子数量的增多,分子间相互作用增强,导致吸附等温线拟合Langmuir吸附等温方程产生偏差. 在不同吸附量下,苯酚在冰表面的吸附构型也表现出不同特征. 吸附量少时,苯酚分子并不倾向于与冰表面平行,而是与冰表面形成约66°的倾斜角;当吸附量增加后,苯酚在冰表面的吸附构型会发生改变. 苯酚在冰表面的吸附能分析结果证实了这一现象,随着苯酚吸附量的增加,苯酚分子间相互作用增加,影响了苯酚在冰表面的吸附方向. 在不同吸附量情况下,苯酚与冰表面形成的主导氢键类型是苯酚的—OH上的H原子与冰表面上的O原子形成的氢键. 并且,在吸附量较多时,平行排布在冰表面的苯酚分子与冰表面水分子之间形成O—H···π键. 本文通过多手段研究结果的互相印证,进一步支持了苯酚力场改进的可行性和吸附结果的准确性. 综上,本文从分子层面探讨了苯酚在冰表面的吸附机理,结果对研究苯酚在气/冰界面的进一步迁移转化奠定了基础,对于增强不同取代基影响芳香类污染物在冰表面吸附机制的理解具有重要意义.
分子模拟研究苯酚在大气冰表面的吸附行为
Molecular simulation study on adsorption behavior of phenol on atmospheric ice surface
-
摘要: 苯酚是大气中重要的有毒污染物,然而其在大气冰表面的吸附行为仍不明确. 本研究使用巨正则蒙特卡罗(GCMC)模拟方法,基于TIP5P水力场和优化的苯酚力场,模拟了温度200 K条件下,苯酚在六边形(Ih)冰0001表面的吸附行为. 研究表明,苯酚在冰表面的吸附等温线具有两个平台期,达到第一个平台期后,苯酚单层吸附饱和并进入快速增长期,随后至吸附凝结点,达到第二个平台期. 当苯酚分子吸附量较小时,苯酚独立地吸附在冰表面,吸附行为符合Langmuir吸附类型;随着苯酚分子吸附量增加,冰表面的苯酚分子间产生相互作用,导致偏离了Langmuir吸附等温线. 在不同吸附量情况下,苯酚与冰表面分子形成的氢键类型均是以苯酚—OH上的氢原子与冰表面氧原子之间形成的氢键为主.
-
关键词:
- 苯酚 /
- 冰表面 /
- 吸附行为 /
- 巨正则蒙特卡罗模拟.
Abstract: Phenol is an important toxic pollutant in the atmosphere, however, its adsorption behavior on the surface of atmospheric ice is still unclear. Here, we employed Grand Canonical Monte Carlo simulations to characterize the adsorption behavior of phenol on hexagonal ice (Ih) at 200 K using the TIP5P water force field and optimized force field of phenol. The results indicate that the adsorption isotherm of phenol on the ice surface has two plateaus. The first plateau corresponds to a saturated monolayer of phenol molecules at the ice surface. After that, the isotherm exhibits a rapid increase to the condensation point, and then reaches the second plateau. At low coverage of phenol on the ice surface, the adsorbed phenol molecules are isolated from each other, and the adsorption isotherm follows the Langmuir adsorption isotherm. With increasing coverage of absorbed phenol molecules, its adsorption isotherms deviated from Langmuir adsorption isotherm due to the interaction between phenol molecules. The type of hydrogen bond formed by phenol molecules and ice surface is dominated by the hydrogen bond formed by the H—atom on the —OH of phenol and the O—atom on the ice surface under different adsorption amounts.-
Key words:
- phenol /
- ice surface /
- adsorption behavior /
- Grand Canonical Monte Carlo simulation.
-

-
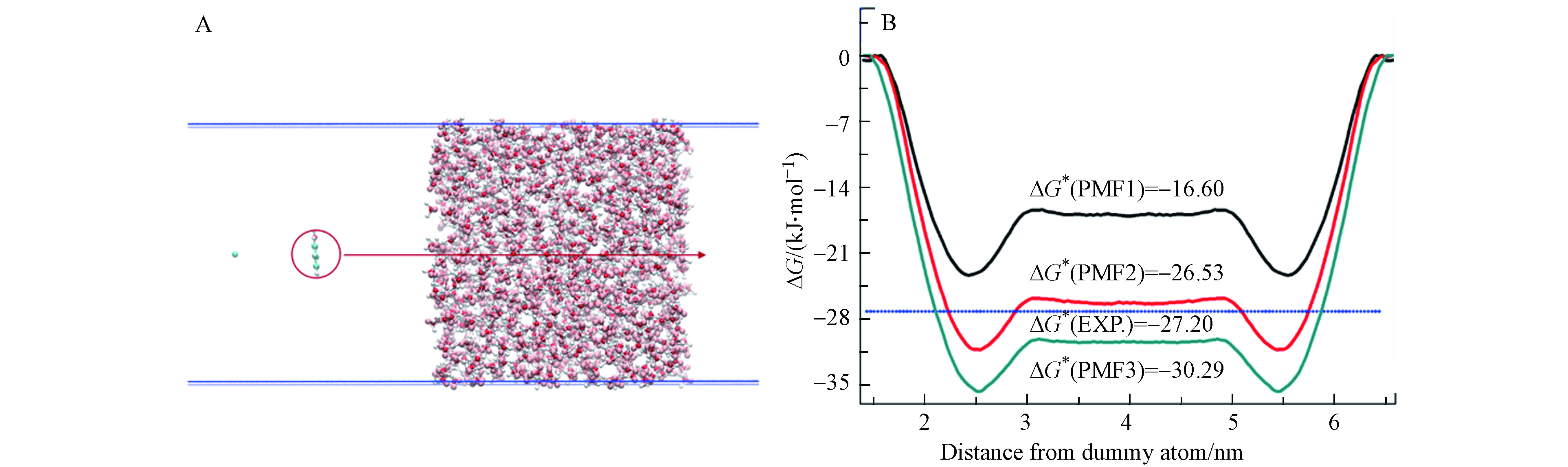
图 1 (A)自由能计算模拟体系:1个苯酚分子、 含有970 个水分子的液相和1个虚原子;(B)不同力场条件下,苯酚分子穿过气水夹层的自由能分布曲线(PMF)
Figure 1. (A) The simulation system for free energy profile: a phenol molecule, a liquid slab and a dummy atom; (B)Potential of mean force (PMF) for moving phenol with different force field parameters through a water slab
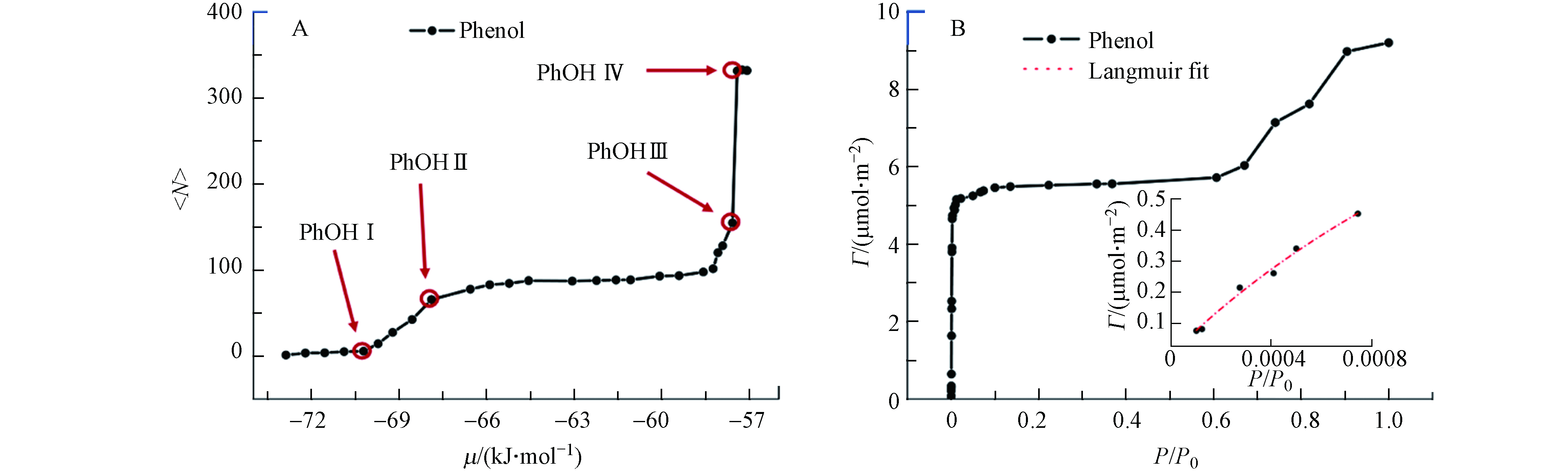
图 2 苯酚两种形式的吸附等温线(A)吸附在冰表面的苯酚分子平均个数
$\left\langle {{N}} \right\rangle $ 与化学势μ的函数;(B)吸附的苯酚分子表面密度与相对压力的函数. 图2(B)的插图显示了低表面密度下与Langmuir吸附等温线拟合的吸附等温线Figure 2. Two forms of phenol adsorption isotherms (A) Average number of phenol molecules adsorbed on the ice surface
$\left\langle {{N}} \right\rangle $ as a function of chemical potential μ; (B) Surface density of adsorbed phenol molecules as a function of relative pressure. The inset of Fig. 2(B) shows the fitting of the adsorption isotherm at low surface density to the Langmuir adsorption isotherm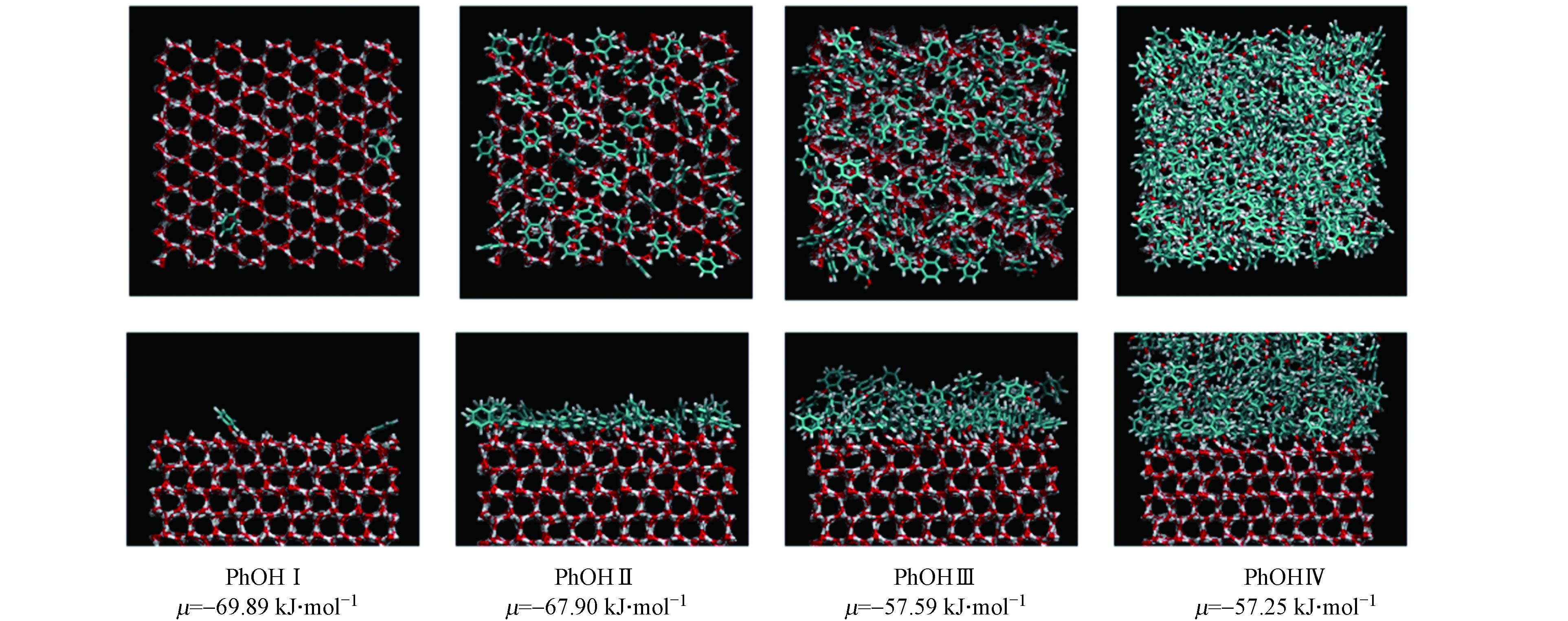
图 3 在选定化学势下,冰表面苯酚吸附层的瞬时平衡快照的俯视图和侧视图
Figure 3. The top and side view of instantaneous equilibrium snapshots for the adsorption layer of phenol on the ice surface at selected μ values

图 4 (A)PhOH Ⅰ—Ⅳ中吸附的苯酚分子中—OH相连的ipso碳原子沿表面X轴位置的密度分布,(B)PhOH Ⅰ—Ⅲ中苯酚分子在冰表面上的吸附方向分布.
Figure 4. (A) The density profiles of the ipso carbon atom linked to—OH of the adsorbed phenol molecules in systems PhOH Ⅰ—Ⅳ along the surface normal axis X and (B) the orientation profiles of the adsorbed phenol molecules at the ice surface in systems PhOH Ⅰ—Ⅲ.
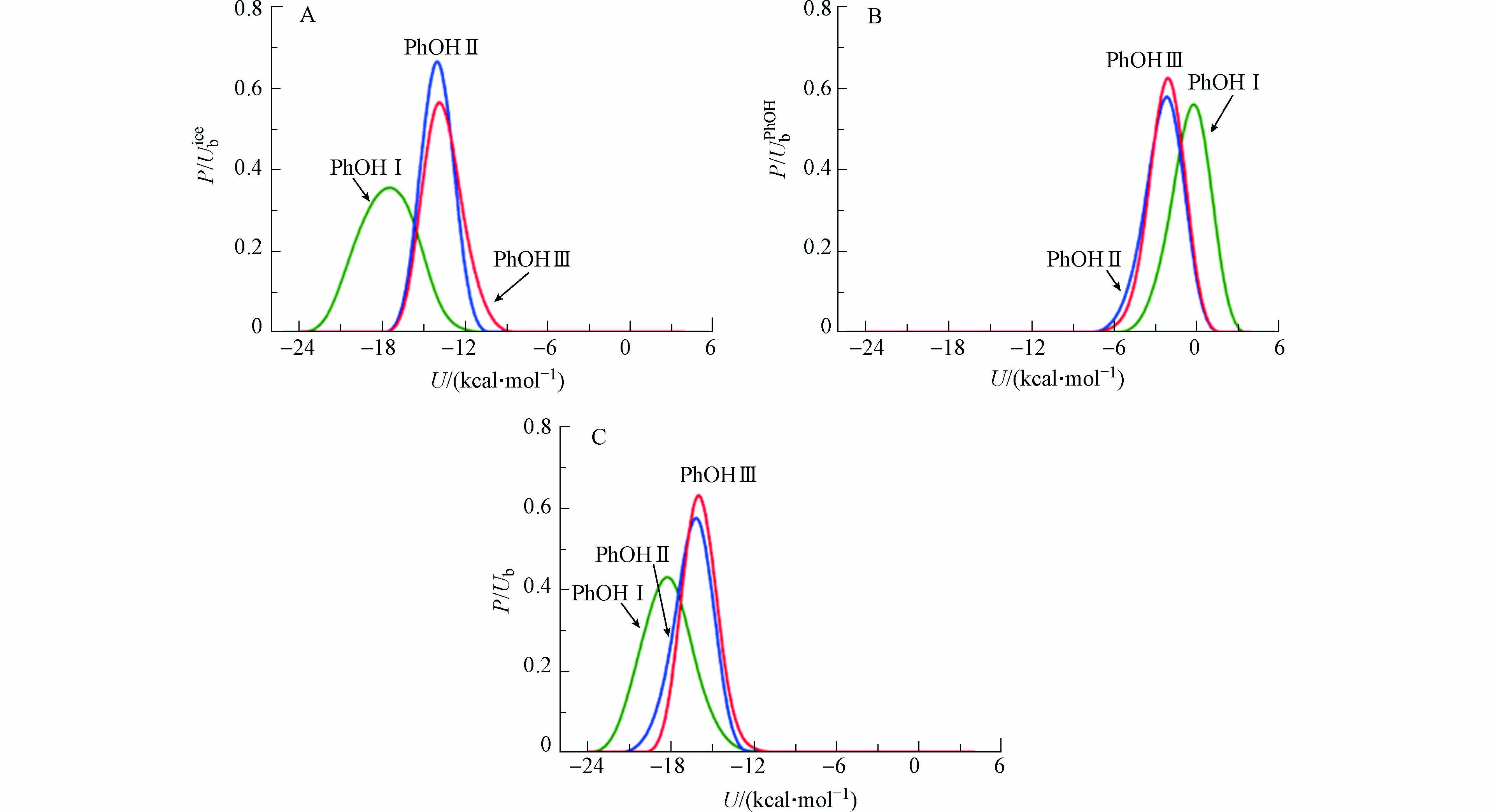
图 5 体系PhOH Ⅰ—Ⅲ中(A)苯酚与冰表面之间的相互作用能(
$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ ), (B)苯酚分子之间相互作用能($ {U}_{\mathrm{b}}^{\mathrm{P}\mathrm{h}\mathrm{O}\mathrm{H}} $ ), (C)苯酚分子与冰表面的总结合能($ {U}_{\mathrm{b}} $ )Figure 5. (A) Distribution of the interaction energy with the ice surface (
$ {U}_{\mathrm{b}}^{\mathrm{i}\mathrm{c}\mathrm{e}} $ ) for the adsorption of phenol, (B) the phenol interaction energy with other adsorbed molecules ($ {U}_{\mathrm{b}}^{\mathrm{P}\mathrm{h}\mathrm{O}\mathrm{H}} $ ), (C) distribution of total binding energy ($ {U}_{\mathrm{b}} $ ) of adsorbed molecule on the ice surface at PhOH Ⅰ—Ⅲ systems, respectively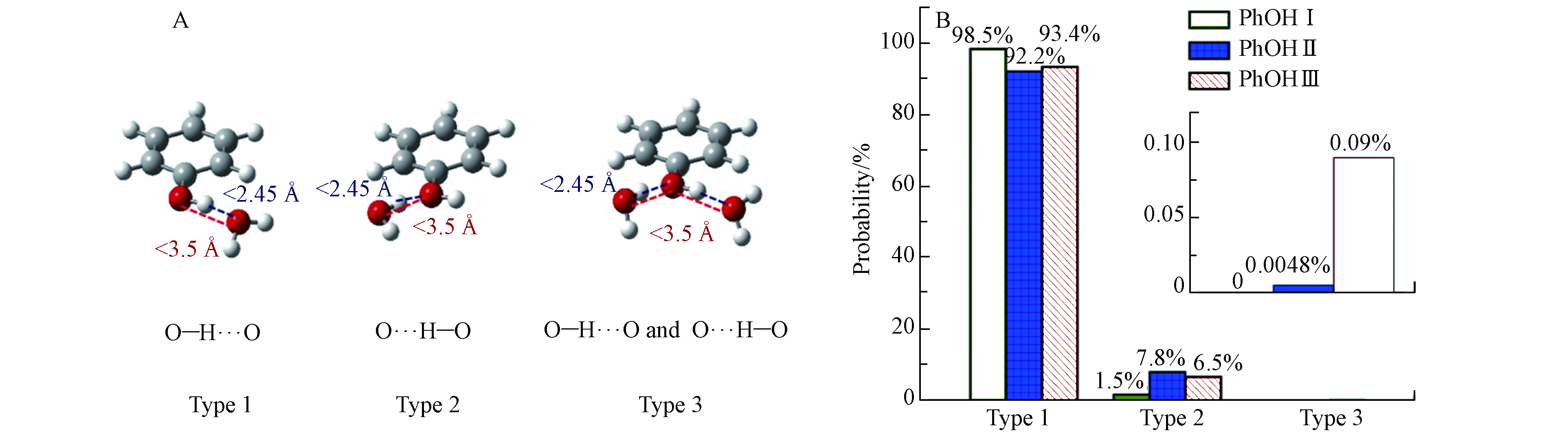
图 6 (A)苯酚的—OH在冰表面形成3种氢键类型,分别命名为“Type1”,“Type2”和“Type3”. (B)苯酚的—OH与冰表面形成3种氢键类型的概率分布
Figure 6. (A) Three types of hydrogen bond formed between—OH of phenol and the ice surface, which are named “Type1”, “Type2” and “Type3”, respectively. (B) Profiles of probability of three types of hydrogen bond formed between —OH of phenol and the ice surface
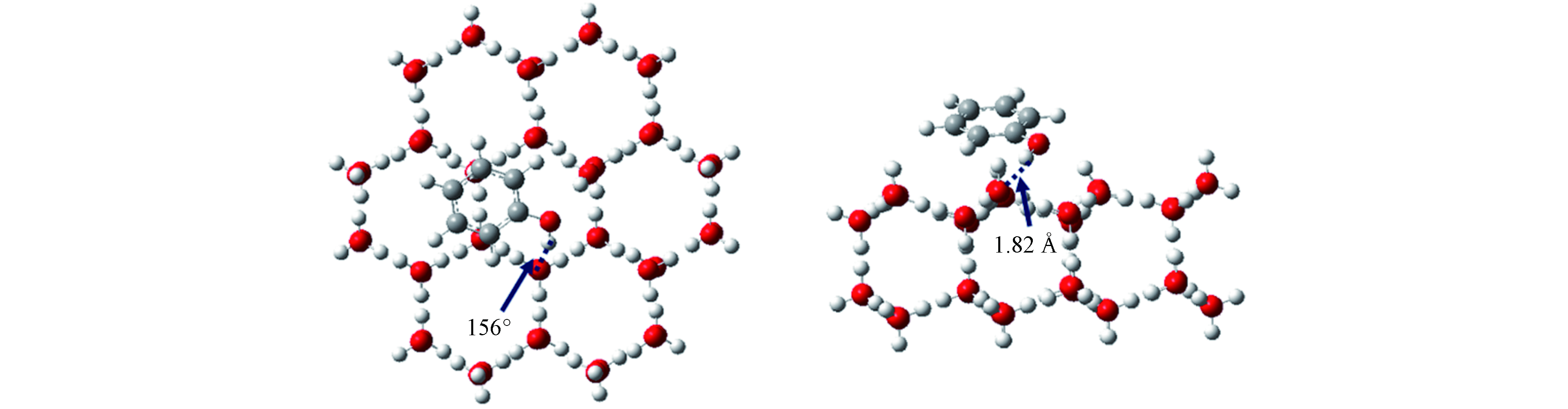
图 7 通过量子化学计算得到苯酚吸附在冰表面的最稳定构型
Figure 7. The most stable adsorption configuration of phenol on ice surface obtained by quantum chemical calculation
表 1 OPLS-AA 力场中苯酚的L-J势能参数和优化后苯酚的原子电荷
Table 1. Lennard-Jones (L-J) Parameters for Phenol Adopted from the OPLS-AA Force Field and Modified Atomic Charges of phenol
苯酚
Phenol原子名称
Atomσ/nm ε/(kcal·mol−1) q/e 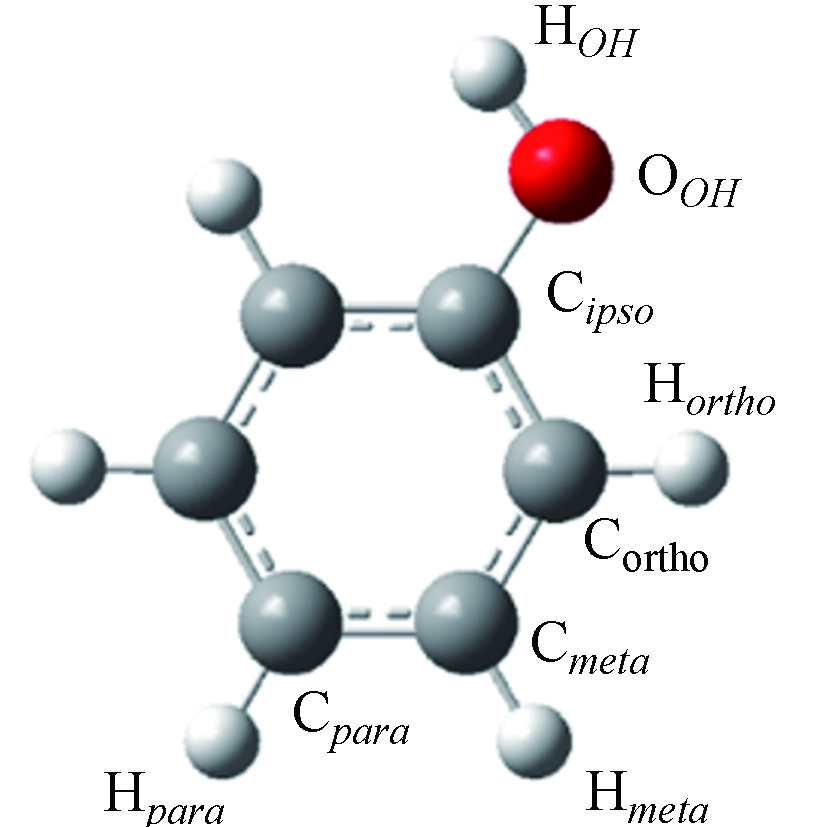
Cipso 0.355 0.070 0.481 Cortho 0.355 0.070 −0.368 Cmeta 0.355 0.070 −0.100 Cpara 0.355 0.070 −0.237 Hortho 0.242 0.030 0.202 Hmeta 0.242 0.030 0.163 Hpara 0.242 0.030 0.163 OOH 0.312 0.170 −0.635 HOH 0.000 0.000 0.434  下载: 导出CSV
下载: 导出CSV
表 2 基于OPLS-AA力场,优化力场下的苯酚偶极矩、密度、蒸发焓和热容计算值与实验值
Table 2. Calculated dipole moment(Debye), density(g·cm−3), evaporation enthalpy(kJ· mol−1) and heat capacity(cal·mol−1·K−1)of phenol with OPLS-AA force field, modified force field and experimental values for phenol
下载: 导出CSV
表 3 苯酚吸附等温线计算数据
Table 3. Calculated data of the phenol adsorption isotherm
化学势
/(kJ·mol−1)
μ吸附个数
N相对压力
Prel表面密度
/(μmol·m−2)
Γ化学势
/(kJ·mol−1)
μ吸附个数
N相对压力
Prel表面密度
/(μmol·m−2)
Γ−72.88 1.27 1.01 $ \times $ 0.08 −63.24 87.11 3.34 $ \times $ 5.18 −72.55 1.36 1.23 $ \times $ 0.08 −63.07 87.28 3.69 $ \times $ 5.19 −72.22 3.58 1.51 $ \times $ 0.21 −62.57 88.38 4.98 $ \times $ 5.25 −71.89 4.45 1.84 $ \times $ 0.26 −62.24 87.84 6.08 $ \times $ 5.22 −71.55 3.83 2.25 $ \times $ 0.23 −61.58 88.51 9.07 $ \times $ 5.26 −71.22 3.61 2.75 $ \times $ 0.21 −61.08 88.62 0.12 5.27 −70.89 5.19 3.35 $ \times $ 0.31 −60.75 88.04 0.15 5.23 −70.56 4.38 4.10 $ \times $ 0.26 −60.41 93.56 0.18 5.56 −70.22 5.72 5.00 $ \times $ 0.34 −60.08 92.99 0.22 5.53 −69.89a 10.87 6.11 $ \times $ 0.65 −59.91 92.26 0.25 5.48 −69.23 27.54 9.12 $ \times $ 1.64 −59.42 93.54 0.33 5.56 −68.56 42.60 1.36 $ \times $ 2.53 −59.25 93.58 0.37 5.56 −67.90b 65.79 2.03 $ \times $ 3.91 −58.75 95.64 0.50 5.69 −67.56 78.38 2.48 $ \times $ 4.66 −58.58 97.70 0.55 5.81 −67.23 79.76 3.03 $ \times $ 4.74 −58.25 101.59 0.67 6.04 −66.57 77.93 4.52 $ \times $ 4.63 −58.08 120.24 0.74 7.15 −66.23 77.84 5.52 $ \times $ 4.63 −57.92 128.29 0.82 7.63 −65.90 83.05 6.74 $ \times $ 4.94 −57.75 151.14 0.90 8.98 −65.57 82.20 8.23 $ \times $ 4.89 −57.59c 154.95 1.00 9.21 −65.23 84.56 1.01 $ \times $ 5.03 −57.42 331.68 −64.90 86.78 1.23 $ \times $ 5.16 −57.25d 332.83 −64.57 87.73 1.50 $ \times $ 5.22 −57.09 332.02 −63.90 87.17 2.24 $ \times $ 5.18 −56.92 330.20 注:a、b、c、d分别对应 PhOH ⅠI、PhOH ⅡII、PhOH Ⅲ 和 PhOH Ⅳ.
下载: 导出CSV
-
[1] BARTELS-RAUSCH T. Chemistry: Ten things we need to know about ice and snow [J]. Nature, 2013, 494(7435): 27-29. doi: 10.1038/494027a [2] BARTELS-RAUSCH T, JACOBI H W, KAHAN T F, et al. Relationship between snow microstructure and physical and chemical processes [J]. Atmospheric Chemistry and Physics, 2012, 12: 30409-30541. [3] HUTHWELKER T, AMMANN M, PETER T. The uptake of acidic gases on ice [J]. Chemical Reviews, 2006, 106(4): 1375-1444. doi: 10.1021/cr020506v [4] ABBATT J P D. Interactions of atmospheric trace gases with ice surfaces: Adsorption and reaction [J]. Chemical Reviews, 2003, 103(12): 4783-4800. doi: 10.1021/cr0206418 [5] DOMINÉ F, CABANES A, LEGAGNEUX L. Structure, microphysics, and surface area of the Arctic snowpack near Alert during the ALERT 2000 campaign [J]. Atmospheric Environment, 2002, 36(15/16): 2753-2765. [6] BARTELS-RAUSCH T, JACOBI H W, KAHAN T F, et al. A review of air–ice chemical and physical interactions (AICI): liquids, quasi-liquids, and solids in snow [J]. Atmospheric Chemistry and Physics, 2014, 14(3): 1587-1633. doi: 10.5194/acp-14-1587-2014 [7] KAHAN T F, ZHAO R, DONALDSON D J. Hydroxyl radical reactivity at the air-ice interface [J]. Atmospheric Chemistry and Physics, 2010, 10(2): 843-854. doi: 10.5194/acp-10-843-2010 [8] DOMINÉ F, SHEPSON P B. Air-snow interactions and atmospheric chemistry [J]. Science, 2002, 297(5586): 1506-1510. doi: 10.1126/science.1074610 [9] DALY G L, WANIA F. Simulating the influence of snow on the fate of organic compounds [J]. Environmental Science & Technology, 2004, 38(15): 4176-4186. [10] MCNEILL V F, GRANNAS A M, ABBATT J P D, et al. Organics in environmental ices: Sources, chemistry, and impacts [J]. Atmospheric Chemistry and Physics, 2012, 12(20): 9653-9678. doi: 10.5194/acp-12-9653-2012 [11] ARELLANO L, FERNÁNDEZ P, TATOSOVA J, et al. Long-range transported atmospheric pollutants in snowpacks accumulated at different altitudes in the Tatra Mountains (Slovakia) [J]. Environmental Science & Technology, 2011, 45(21): 9268-9275. [12] GERBER R B, VARNER M E, HAMMERICH A D, et al. Computational studies of atmospherically-relevant chemical reactions in water clusters and on liquid water and ice surfaces [J]. Accounts of Chemical Research, 2015, 48(2): 399-406. doi: 10.1021/ar500431g [13] JEDLOVSZKY P, PÁRTAY L, HOANG P N M, et al. Determination of the adsorption isotherm of methanol on the surface of ice. an experimental and grand canonical Monte Carlo simulation study [J]. Journal of the American Chemical Society, 2006, 128(47): 15300-15309. doi: 10.1021/ja065553+ [14] DARVAS M, PICAUD S, JEDLOVSZKY P. Molecular dynamics simulation of the adsorption of oxalic acid on an ice surface [J]. ChemPhysChem, 2010, 11(18): 3971-3979. doi: 10.1002/cphc.201000513 [15] KISS B, PICAUD S, SZŐRI M, et al. Adsorption of formamide at the surface of amorphous and crystalline ices under interstellar and tropospheric conditions. A grand canonical Monte Carlo simulation study [J]. The Journal of Physical Chemistry. A, 2019, 123(13): 2935-2948. doi: 10.1021/acs.jpca.9b00850 [16] DARVAS M, LASNE J, LAFFON C, et al. Adsorption of acetaldehyde on ice as seen from computer simulation and infrared spectroscopy measurements [J]. Langmuir:the ACS Journal of Surfaces and Colloids, 2012, 28(9): 4198-4207. doi: 10.1021/la204472k [17] LIU Y, SHAO M, FU L L, et al. Source profiles of volatile organic compounds (VOCs) measured in China: Part I [J]. Atmospheric Environment, 2008, 42(25): 6247-6260. doi: 10.1016/j.atmosenv.2008.01.070 [18] GUO H, SO K L, SIMPSON I J, et al. C1-C8 volatile organic compounds in the atmosphere of Hong Kong: Overview of atmospheric processing and source apportionment [J]. Atmospheric Environment, 2007, 41(7): 1456-1472. doi: 10.1016/j.atmosenv.2006.10.011 [19] MASSOLO L, REHWAGEN M, PORTA A, et al. Indoor-outdoor distribution and risk assessment of volatile organic compounds in the atmosphere of industrial and urban areas [J]. Environmental Toxicology, 2010, 25(4): 339-349. [20] CAZIER F, GENEVRAY P, DEWAELE D, et al. Characterisation and seasonal variations of particles in the atmosphere of rural, urban and industrial areas: Organic compounds [J]. Journal of Environmental Sciences, 2016, 44: 45-56. doi: 10.1016/j.jes.2016.01.014 [21] MÉSZÁR Z E, HANTAL G, PICAUD S, et al. Adsorption of aromatic hydrocarbon molecules at the surface of ice, as seen by grand canonical Monte Carlo simulation [J]. The Journal of Physical Chemistry C, 2013, 117(13): 6719-6729. doi: 10.1021/jp401532x [22] PETITJEAN M, HANTAL G, CHAUVIN C, et al. Adsorption of benzaldehyde at the surface of ice, studied by experimental method and computer simulation [J]. Langmuir, 2010, 26(12): 9596-9606. doi: 10.1021/la100169h [23] FU Z H, HE N, ZHOU P T, et al. Grand canonical Monte Carlo simulation on adsorption of aniline on the ice surface [J]. Journal of Molecular Liquids, 2019, 290: 111221. doi: 10.1016/j.molliq.2019.111221 [24] FU Z H, HE N, ZHOU P T, et al. Adsorption of nitrobenzene on the surface of ice: A grand canonical Monte Carlo simulation study [J]. The Journal of Physical Chemistry C, 2017, 121(29): 15746-15755. doi: 10.1021/acs.jpcc.7b03531 [25] XU C, WANG L M. Atmospheric oxidation mechanism of phenol initiated by OH radical [J]. The Journal of Physical Chemistry. A, 2013, 117(11): 2358-2364. doi: 10.1021/jp308856b [26] NOLTE C G, SCHAUER J J, CASS G R, et al. Highly polar organic compounds present in wood smoke and in the ambient atmosphere [J]. Environmental Science & Technology, 2001, 35(10): 1912-1919. [27] SCHAUER J J, KLEEMAN M J, CASS G R, et al. Measurement of emissions from air pollution sources. 5. C1−C32 organic compounds from gasoline-powered motor vehicles [J]. Environmental Science & Technology, 2002, 36(6): 1169-1180. [28] BRODZINSKY R, SINGH H B. Volatile organic chemicals in the atmosphere: An assessment of available data[M]. US Environmental Protection Agency, Environmental Sciences Research Laboratory, 1983. [29] BONONI F C, CHEN Z K, ROCCA D, et al. Bathochromic shift in the UV-visible absorption spectra of phenols at ice surfaces: Insights from first-principles calculations [J]. The Journal of Physical Chemistry. A, 2020, 124(44): 9288-9298. doi: 10.1021/acs.jpca.0c07038 [30] SUMI I, FÁBIÁN B, PICAUD S, et al. Adsorption of fluorinated methane derivatives at the surface of ice under tropospheric conditions, as seen from grand canonical Monte Carlo simulations [J]. The Journal of Physical Chemistry C, 2016, 120(31): 17386-17399. doi: 10.1021/acs.jpcc.6b04300 [31] RICK S W. A reoptimization of the five-site water potential (TIP5P) for use with Ewald sums [J]. The Journal of Chemical Physics, 2004, 120(13): 6085-6093. doi: 10.1063/1.1652434 [32] ABASCAL J L F, VEGA C. A general purpose model for the condensed phases of water: TIP4P/2005 [J]. The Journal of Chemical Physics, 2005, 123(23): 234505. doi: 10.1063/1.2121687 [33] MAHONEY M W, JORGENSEN W L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions [J]. The Journal of Chemical Physics, 2000, 112(20): 8910-8922. doi: 10.1063/1.481505 [34] CALEMAN C, van MAAREN P J, HONG M Y, et al. Force field benchmark of organic liquids: Density, enthalpy of vaporization, heat capacities, surface tension, isothermal compressibility, volumetric expansion coefficient, and dielectric constant [J]. Journal of Chemical Theory and Computation, 2012, 8(1): 61-74. doi: 10.1021/ct200731v [35] JORGENSEN W L, LAIRD E R, NGUYEN T B, et al. Monte Carlo simulations of pure liquid substituted benzenes with OPLS potential functions [J]. Journal of Computational Chemistry, 1993, 14(2): 206-215. doi: 10.1002/jcc.540140208 [36] PRICE M L P, OSTROVSKY D, JORGENSEN W L. Gas-phase and liquid-state properties of esters, nitriles, and nitro compounds with the OPLS-AA force field [J]. Journal of Computational Chemistry, 2001, 22(13): 1340-1352. doi: 10.1002/jcc.1092 [37] VÁCHA R, JUNGWIRTH P, CHEN J, et al. Adsorption of polycyclic aromatic hydrocarbons at the air-water interface: Molecular dynamics simulations and experimental atmospheric observations [J]. Physical Chemistry Chemical Physics:PCCP, 2006, 8(38): 4461-4467. doi: 10.1039/B610253K [38] VÁCHA R, SLAVÍČEK P, MUCHA M, et al. Adsorption of atmospherically relevant gases at the air/water interface: Free energy profiles of aqueous solvation of N2, O2, O3, OH, H2O, HO2, and H2O2 [J]. The Journal of Physical Chemistry A, 2004, 108(52): 11573-11579. doi: 10.1021/jp046268k [39] HESS B, KUTZNER C, van der SPOEL D, et al. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation [J]. Journal of Chemical Theory and Computation, 2008, 4(3): 435-447. doi: 10.1021/ct700301q [40] HIRSCH T K, OJAMÄE L. Quantum-chemical and force-field investigations of ice ih: Computation of proton-ordered structures and prediction of their lattice energies [J]. The Journal of Physical Chemistry B, 2004, 108(40): 15856-15864. doi: 10.1021/jp048434u [41] BOCKSTEDTE M, MICHL A, KOLB M, et al. Incomplete bilayer termination of the ice (0001) surface [J]. The Journal of Physical Chemistry C, 2016, 120(2): 1097-1109. doi: 10.1021/acs.jpcc.5b10836 [42] NETZ R R, HORINEK D. Progress in modeling of ion effects at the vapor/water interface [J]. Annual Review of Physical Chemistry, 2012, 63: 401-418. doi: 10.1146/annurev-physchem-032511-143813 [43] BUCH V, SIGURD B, PAUL DEVLIN J, et al. Solid water clusters in the size range of tens-thousands of H2O: A combined computational/spectroscopic outlook [J]. International Reviews in Physical Chemistry, 2004, 23(3): 375-433. doi: 10.1080/01442350412331316124 [44] HONTI B, SZŐRI M, JEDLOVSZKY P. Description of the interfacial behavior of benzonitrile at icy surfaces by grand canonical Monte Carlo simulations [J]. The Journal of Physical Chemistry. A, 2022, 126(7): 1221-1232. doi: 10.1021/acs.jpca.1c10749 [45] SÁNCHEZ M A, KLING T, ISHIYAMA T, et al. Experimental and theoretical evidence for bilayer-by-bilayer surface melting of crystalline ice [J]. Proceedings of the National Academy of Sciences of the United States of America, 2017, 114(2): 227-232. doi: 10.1073/pnas.1612893114 [46] KLING T, KLING F, DONADIO D. Structure and dynamics of the quasi-liquid layer at the surface of ice from molecular simulations [J]. The Journal of Physical Chemistry C, 2018, 122(43): 24780-24787. doi: 10.1021/acs.jpcc.8b07724 [47] PICAUD S, JEDLOVSZKY P. Molecular-scale simulations of organic compounds on ice: Application to atmospheric and interstellar sciences [J]. Molecular Simulation, 2019, 45(4/5): 403-416. [48] MEZEI M. MMC: Monte Carlo program for simulation of molecular assemblies [EB/OL]. [2020-07-10] [49] MEZEI M. A cavity-biased (T, V, μ) Monte Carlo method for the computer simulation of fluids [J]. Molecular Physics, 1980, 40(4): 901-906. doi: 10.1080/00268978000101971 [50] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09 [CP]. Wallingford, CT: Gaussian, Inc, 2009. [51] SANDER R. Compilation of Henry's law constants (version 4.0) for water as solvent [J]. Atmospheric Chemistry and Physics, 2015, 15(8): 4399-4981. doi: 10.5194/acp-15-4399-2015 [52] O'NIEL M J. The merck index: An encyclopedia of chemicals, drugs, and bilogicals, (13th ed)[M]. Whitehouse station, NJ: Merck and Co. , Inc. , 2001. [53] CHICKOS J S, HOSSEINI S, HESSE D G. Determination of vaporization enthalpies of simple organic molecules by correlations of changes in gas chromatographic net retention times [J]. Thermochimica Acta, 1995, 249: 41-62. doi: 10.1016/0040-6031(95)90670-3 [54] KUDCHADKER S A, KUDCHADKER A P, WILHOIT R C, et al. Ideal gas thermodynamic properties of phenol and cresols [J]. Journal of Physical and Chemical Reference Data, 1978, 7(2): 417-423. doi: 10.1063/1.555573 [55] PAGLIAI M, CARDINI G, RIGHINI R, et al. Hydrogen bond dynamics in liquid methanol [J]. The Journal of Chemical Physics, 2003, 119(13): 6655-6662. doi: 10.1063/1.1605093 [56] da SILVA J A, MOREIRA F G, dos SANTOS V M L, et al. Hydrogen bond networks in water and methanol with varying interaction strengths [J]. Physical Chemistry Chemical Physics:PCCP, 2011, 13(2): 593-603. doi: 10.1039/C0CP01204A [57] ZHANG N, RUAN X H, SONG Y C, et al. Molecular dynamics simulation of the hydration structure and hydrogen bonding behavior of phenol in aqueous solution [J]. Journal of Molecular Liquids, 2016, 221: 942-948. doi: 10.1016/j.molliq.2016.06.048 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 3532
- HTML全文浏览数: 3532
- PDF下载数: 111
- 施引文献: 0
















