


 下载:
下载:
-
代谢组这一术语于1998年提出[1],是细胞或生物体中参与代谢反应的小分子化合物的集合. 类似于基因组学、转录组学和蛋白质组学,代谢组学是系统生物学的一门“组学”科学,旨在对生物体中存在的内源性和外源性代谢物进行分析[2]. 代谢组学的研究对象涵盖了各种不同极性的化合物,这些化合物具有广泛的化学类别,且浓度变化范围达到9个数量级[3],因此如何提升代谢物分析方法的覆盖率以更全面的识别代谢物是代谢组学的首要挑战之一. 高覆盖代谢组学可以同时进行大量代谢物的定性定量分析,并探究与生物表型的因果相关性,识别潜在的生物标志物[4-5],因此高覆盖代谢组学也是评估外暴露引起的生物毒性效应的重要方法,将高覆盖代谢组学方法用于环境毒理学中可以识别污染物暴露引起的代谢物变化,研究污染物与表型之间的毒性效应机制.
高覆盖代谢组学分析的主要流程包括样品前处理,仪器分析和数据分析. 目前已经有许多研究针对代谢物提取方法进行开发与优化[6-10]. 另外随着技术进步,仪器分析平台也更多样化,包括分离技术平台(例如液相色谱(LC)、气相色谱(GC)和毛细管电泳(CE)等)和检测技术平台(例如质谱(MS)和核磁共振(NMR)). 其中液相色谱-质谱联用(LC-MS)仪器在引入电喷雾电离(ESI)后提高了灵敏度,促进了组学规模的代谢物检测[11-12],已经成为代谢组学的重要仪器分析平台. 在数据分析方面,已经开发了更全面的代谢物数据库[13-14],助力代谢物的识别与发现. 计算机的最新技术和生物信息学工具飞速发展,提升了代谢组学数据的处理速度,并将继续推动代谢组学领域的发展. 然而,先进的数据处理工具也无法分析未被仪器采集到的代谢物数据,因此,仪器分析方法的选择对代谢物数据的全面采集和后续的数据分析是十分重要的.
LC-MS平台在进行仪器分析时,代谢物首先以不同的保留时间(RT)经LC洗脱后,进入MS采集母离子的一级谱图(MS1),之后对所选择的母离子进行碎裂采集二级碎片谱图(MS/MS),二级碎片谱图是代谢物结构鉴定的关键. 因此,在基于质谱的仪器分析方法中,数据采集策略决定了如何选择母离子以进一步裂解,是影响用于后续分析的MS/MS的覆盖范围和质量的重要因素.
现有的相关研究多是对基于质谱的代谢组学分析方法的全过程概述,包括了从色谱分离方法到质谱采集以及数据处理的全过程[15-16],但当前还没有聚焦于质谱数据采集策略研究进展的最新综述,本文将补充此方向的研究空白,重点综述2015年以来基于LC-MS的高覆盖代谢组学数据采集策略的研究进展. 当前应用于代谢组学的质谱分析方法包括靶向、非靶向以及两者结合的混合方法,本文将综述针对这3种方法开发的数据采集策略的进展,并介绍高覆盖代谢组学在环境毒理学领域的最新应用.
-
靶向代谢组学方法是对已知代谢物进行定性和定量的方法,当前靶向代谢组学常用的数据采集策略主要包括多反应监测(MRM)和平行反应监测(PRM).
-
基于三重四极杆质谱(QQQ)的多反应监测(MRM)是靶向代谢组学中应用最广泛的数据采集策略,被称为小分子和代谢物定量的金标准[17-18]. 在用MRM进行代谢物分析前,需要先通过标准品对目标代谢物的质谱参数(碰撞能和去簇电压)和离子对信息(前体-产物离子对)进行优化,选择最优灵敏度. 在MRM采集时,Q1选择感兴趣的目标母离子,Q2作为碰撞池对目标前体进行碎裂,Q3对预选的子离子进行分离(方法流程图可见下图1). 虽然方法建立之初需要对每个目标物进行参数优化,但该方法定量灵敏度和准确性很高,可以对极少量的样品或极低浓度的样品进行准确的定量分析,如有研究用10 μL全血对36种目标代谢物进行靶向分析,结果呈现优异的稳定性和可重复性[19]. 然而由于依赖于标准品,这种传统MRM方法的代谢物覆盖范围有限.
目前,已经开发了新的方法提升基于MRM的靶向代谢组学覆盖率. 基于合成大量标准品进行靶向分析的靶向试剂盒技术增加了目标代谢物的覆盖范围,该方法使基于MRM的靶向方法可以实现快速定量数百种目标代谢物,例如有研究使用AbsoluteIDQ™ p180 kit代谢组试剂盒进行靶向代谢组分析,可靠鉴定了159种血清代谢物[20];而一些靶向代谢组学试剂盒能对500多种化合物进行检测量化[21-22]. 虽然方法得到了很好的优化,但这种多反应监测方法仍然依赖于目标物的标准品. 为了摆脱对标准品的依赖,进一步增加多反应监测方法的覆盖度,Domingo-Almenara等[23]建立了用于小分子靶向分析的MRM离子对库,该数据库包含了超过15500个分子的实验或计算机生成MRM离子对,可以实现大量内源性和外源性代谢物的靶向识别与量化. 这种基于建立的MRM数据库进行样品MRM采集的方法可以分析没有标准品的物质,提高分析覆盖度,但不同仪器或样本类型之间的兼容性通常不好[24].
-
高分辨率质谱仪(HRMS)的发展为化合物分析提供了新方法. 基于HRMS进行的靶向数据采集,常称为平行反应监测(PRM),也被描述为高分辨MRM[25]. 其工作流程如图1所示,与MRM类似,PRM采集模式同样是在Q1进行母离子的筛选,在Q2进行母离子碎裂,不同之处在于,PRM第三步通过高分辨的质谱进行所有碎片离子的测定.
基于HRMS的PRM采集模式能以17500—35000分辨率进行MS分析,可以分辨小于0.01 Da的质量差异,从而可以有效地将目标物与其他干扰离子区分开[25],因此可以采集高准确度MS/MS,识别目标代谢物的离子进行定量,对标准品的依赖较低,这是PRM的一大优势. PRM已经应用于多个生物学领域的靶向肽定量[26-27]. Zhou等[25]在Q-Orbitrap仪器上开发了针对237种极性代谢物的PRM方法,通过在不同种生物样品中进行应用,证明了PRM在大规模靶向代谢物定量方面出色的灵敏度、准确性和适用性. 另外,PRM由于监测了所有二级碎片离子,优化过程更简单,相比MRM在方法开发阶段耗时少. 但鉴于高分辨质谱扫描速度较慢,PRM在同时进行大规模靶向分析方面存在限制. 相较而言,QQQ仪器具有高扫描速度和灵敏度,这缩短了MRM采集的循环时间,有利于更高效的分析,并确保足够的色谱峰数据点进行准确定量[25].
MRM方法优势在于高扫描速度和极性切换,而PRM具有高分辨率和全碎片采集的优点. 在大规模靶向代谢组定量分析时,可以考虑结合两者的优势,将PRM作为MRM的补充工具,先经PRM获得代谢物二级谱图信息,再转为MRM进行高速扫描分析[25].
-
非靶向代谢组学是在没有标准品的情况下无偏向性的对所有采集到的谱图进行检测,以达到高覆盖的非靶向代谢组学分析. 非靶向代谢物鉴定通常依赖于高分辨MS1谱图和MS/MS谱图,其中MS1谱图用于确定分子式,MS/MS谱图用于代谢物结构鉴定,因此首先需要采集全面且准确的代谢物MS1和MS/MS谱图信息. 高分辨率质谱已被证明是非靶向代谢组学分析的强大信息采集工具,因其能获得高分辨率和高质量准确度的扫描信息. 基于高分辨质谱的非靶向代谢组学获取谱图数据主要有两种方式:对用于全局分析的MS1数据和用于结构鉴定的MS/MS数据分别进行采集;或者在一次运行中同时采集MS1和MS/MS数据[28].
-
全扫描(Full scan)是对MS1和MS/MS数据分别进行采集的方法,是代谢组学中常用到的数据采集方法[29-30]. 首先对样品进行全扫描,通过较低的碰撞能量即可获得所有代谢特征的MS1信息. 然后经预处理软件(如XCMS等)提取代谢特征[31],基于给定生物学问题选择感兴趣的特征在后续实验中进行MS/MS采集,鉴定代谢物. 全扫描在采集、数据处理方面比较简单,但它将代谢物定量与结构鉴定分离开来,需要额外的仪器分析时间和样品量[32],并且多次分析之间间隔的时间可能会出现数据难以对齐和样品变化等问题[28].
近年来越来越受到关注的另一种方式是在一次运行中同时采集MS1和MS/MS数据. 这种方法主要有两种模式:数据依赖型扫描(data dependent acquisition,DDA)和数据非依赖型扫描(data independent acquisition,DIA).
-
DDA是在全扫描基础上开发的一种更省时快速的数据采集模式,是目前非靶向分析中常用的方法. DDA模式下,质谱仪自动地在Full scan MS和MS/MS采集间切换,在每个循环中先进行MS全扫描采集一级质谱信息,然后选择目标母离子进行碎裂并获得二级谱图,进行物质鉴定,如图2所示[33]. 目标母离子的筛选标准有离子丰度,信噪比,同位素分布等,最常见的是选择强度最高的前几个离子进行碎裂. DDA在一次分析中同时获得MS1和筛选母离子的MS/MS[32],可以同时进行数据处理和代谢物鉴定. 且DDA采用较窄的m/z窗口筛选目标母离子,减少了干扰离子的存在,可以提供较高质量的MS/MS. 但由于目标母离子的筛选通常取决于强度,一些感兴趣的低丰度母离子不能被碎裂,无法获取二级谱图,导致传统DDA方法难以鉴定低丰度物质,且由于筛选过程存在一定随机性,在每次分析时选取的母离子并不完全相同,导致方法重现性较低[34].
-
(1)基于排除或包含列表的数据依赖型扫描方法
由于传统的DDA方法难以鉴定低丰度物质,已有研究通过对传统DDA方法的改进,提高了方法的覆盖范围和性能. 首先是基于DDA设置排除列表的方法,将背景离子或不需碎裂的离子列入排除列表,在下一次仪器分析时不进行碎裂. 静态排除列表需要首先运行空白样品,以生成背景离子排除列表,这种方法通过过滤无关特征来增加代谢物的二级谱图覆盖率,可以排除基线噪声或背景干扰,但对鬼峰的排除效率较低[35]. 近年来结合多针进样的动态排除列表方法得到发展,这种方法在第一针采用DDA方法进样,将获得的有二级谱图的母离子及其保留时间范围输入排除列表中,使得已采集二级谱图的母离子在下一针进样时不再被选择,从而使更低丰度的母离子可以被选择[35],通过多针进样提高二级谱图覆盖率. 这种方法最早应用于蛋白质组学,Bendall等[36]利用这一原理开发了迭代排除质谱法(IE-MS),与5次重复DDA相比,5次IE-MS识别的独特蛋白质增加了30%,降低了对低丰度肽的数据采集偏差. 动态列表方法也被应用于代谢组学中,Edmands等[37]通过6次重复进样的迭代排除方法对经常食用富含多酚食物的人的尿液进行分析,确定了与所选食物摄入相关的80多种多酚代谢物. 由于手动生成排除列表的工作耗时,Koelmel等[38]开发了R脚本“IE-omics”,输入原始数据即可自动生成排除列表,将其应用于脂质组学可将识别率提高50%以上. 理论上,迭代排除DDA方法可通过不断迭代来获取所有高于背景信号的母离子的二级谱图,但在分析大批量样品时要评估重复进样增加的仪器时间成本. 有研究发现在二级谱图质量和覆盖率之间的最好折衷方案是进行五轮迭代排除,这样即可大大提高二级覆盖率[39],为鉴定提供大量信息.
基于DDA设置包含列表的方法,是通过将样品相关离子列入包含列表,优先选择包含列表中母离子进行碎裂,以增加特异性MS/MS覆盖率[40]. Roger Giné等[41]介绍了一种以分子式为导向的采集方法HERMES,根据分析样品的特异性信息选择独特分子式,在进行二级谱图扫描时选择包含列表中的这些样品相关前体离子,从而提高代谢物分析的选择性和二级谱图的覆盖度,结果表明HERMES鉴定的代谢物是三轮迭代排除DDA方法的两倍.
目前也有研究将包含列表与排除列表相结合进一步增加二级谱图覆盖范围. Cho等[28]基于空白扣除过滤非生物特征,基于实际样品分析生成包含独特生物代谢物的离子列表,集成了排除列表和包含列表,这种方法在四次进样后获取了>99%的独特代谢物的二级谱图,显著提高了覆盖率. 近年来也提出了更加自动化的手段推动了方法的应用. AcquireX智能化数据采集模式[42]基于机器学习的方法自动迭代更新背景排除和前体包含列表,在同一样品重复进样中,可以实现相关特征的高覆盖DDA采集.
(2)数据依赖型扫描方法的智能化改进
除了通过排除和包含列表进行DDA方法的改进,目前还开发了其他智能化手段进行DDA方法的改进. Broeckling等[43]考虑到代谢组学数据采集建立在大队列的样品组上,提出了一种数据集依赖型采集模式(DsDA),通过实时分析已采集数据,及时优化后续序列中MS/MS采集方案,并利用R等软件实现了自动化数据分析. 通过在数据处理和数据采集之间的近乎实时反馈循环,可以获得全面的二级覆盖范围. 最近,Bills等[44]建立了一种新的采集策略,称为Met-IQ,将实验MS/MS实时地与参考数据库比较,谱图相似则自动执行多级质谱(MSn)采集,获得更多结构信息,通过将MS3采集限制在可能的目标化合物上,从而减少了MS3的触发,提高了MS2采集率. Wandy等[45]介绍了一种虚拟代谢组学质谱仪(ViMMS),是一种用于代谢组学的模块化LC-MS/MS模拟器,可以对不同策略进行测试并获得模拟结果,允许在计算机上对二级采集进行优化而无需占用仪器时间. Davies等[46]扩展了ViMMS的使用,通过仪器应用程序编程接口(IAPI)创建了允许ViMMS开发的控制器直接在实际质谱上使用的桥接代码. 用这种方法开发了两种新的DDA策略并应用于实际代谢物分析,结果均优于传统DDA的覆盖范围. 这种基于计算机模拟、优化和验证的过程为未来的采集策略改进提供了方向.
这些方法的改进在不同程度上提高了DDA的二级覆盖率,并通过与可用数据库的匹配进行鉴定. 如Chen等[47]在Q-TOF仪器上开发了一种DDA方法研究人类泪液代谢组,将MS/MS谱图与METLIN[48]、HMDB[14]和Massbank[49]数据库进行匹配,鉴定出60种代谢物. 所有这些研究为集成HRMS和DDA工作流程以识别、鉴定和量化生物提取物中的代谢特征铺平了道路. 然而,为了更好地弥补由于母离子选择和欠采样带来的限制,可以无差别对每个母离子进行碎裂的数据非依赖型采集(DIA)工作流程逐渐被开发和广泛应用[34].
-
数据非依赖型扫描(data independent acquisition,DIA)下,质谱仪通过高低碰撞能量交替采集信息,低能量时采集一级质谱,高能量时打碎离子无偏获取全部二级碎片信息(图2分别展示了DDA和DIA的工作流程). 传统的DIA方法包括:MSE、MSALL或MS-AIF[35, 50]. Wrona等[51]首先在LC-QTOF上开发了这种数据采集方法,并成功应用于维拉帕米(心血管药物)体外代谢物的鉴定. 之后Plumb等[52]描述了UPLC/MSE的方法,通过高低碰撞能交替获得所有离子MS/MS数据,并在大鼠尿液的内源物分析中验证了该方法. 类似的全离子碎裂(AIF)方法也已经在Orbitrap仪器上开发并应用[53-56]. 与DDA不同,DIA不预先筛选母离子,理论上能获取所有母离子的碎片信息. 然而明显的缺陷在于母离子与其碎片离子之间的联系被解离,所得碎片谱图复杂而无法直接将碎片对应到特定的母离子[57],对后续代谢物的鉴定带来了挑战. 因此需要对传统DIA方法进行改进,同时结合谱图解卷积的软件来应对这一挑战.
-
当前已经开发了多种方法对传统DIA进行改进,例如将离子淌度分析仪(IMS)耦合到DIA工作流程中. IMS通过额外确定碰撞截面(CCS)值(与离子形状相关的参数),可以提高代谢物鉴定的准确性,甚至解决同分异构体的分离问题[58]. Paglia等[58]将离子淌度集成到DIA中,可以分离共流出的L-磷脂酰乙醇胺,证明了在碎片化前进行离子淌度分离能提供更清晰的MS/MS谱图,促进建立母离子和碎片离子之间的联系,从而提高鉴定的特异性. 同时IMS获得的CCS值具有非常高的重现性(RSD<2%)[59].
为了提高特异性,Gillet等[60]引入了另一种改进方法,即基于Q-TOF的所有理论质谱的顺序窗口采集(SWATH),该方法将扫描范围划分为以某一固定宽度(如20或25Da)为间隔的连续区间. Zhang等[61]通过在SWATH采集中使用可变隔离窗口进一步提高了LC-SWATH-MS采集的选择性. 目前SWATH方法已广泛应用于非靶向代谢组学[57, 62-67]. 近年来还提出了其他改进策略进一步提高选择性. Moseley等[68]描述了一种称为SONAR的新型扫描四极杆DIA方法,对质量范围内的母离子进行快速移动的四极杆扫描,根据四极杆移动位置将母离子与子离子相关联,具有更高的选择性. 另有研究开发了改进的ScanningSWATH[69],将SWATH的Q1分段式质量窗口采集转变为按一定窗口大小连续的Q1采集,通过更小的隔离窗口提供更好的谱图专属性. 此外还有通过小隔离窗口获得MS/MS谱图的PASS-DIA方法[70]. 最近有研究提出了一种数据依赖辅助数据独立采集(DaDIA)的新工作流程,对单个样品执行DIA,合并样品进行DDA,结合了DIA的高MS2覆盖率和DDA的高MS2谱图质量[71]. 除上述方法外,还有一些更早的DIA改进策略,可以参见Chapman等于2014年发表的综述[72].
尽管如此,这些改进的DIA技术仍不能完全可靠地建立母离子和碎片离子之间的联系,需要使用算法软件对嵌合的MS/MS谱图解卷积. 目前已成功开发了一些DIA数据的解卷积软件,如CorrDec、MS-DIAL、DecoMetDIA或MetDIA等[73-76]. 解卷积后的谱图可以与谱库比对鉴定,如今质谱数据库不断扩大,如MassBank[49]、METLIN[77]、HMDB[14]或GNPS[78]等,但数量和可用性仍不够,且扩展这些实验衍生的质谱数据库非常耗时,当前在开发谱图预测和代谢物注释的计算机生成质谱的计算工具方面付出了相当大的努力.
总的来说,靶向方法旨在识别和量化有限数量的已知代谢物,具有可靠的定量准确性、宽动态范围和高灵敏度,但通常无法检测未知物. 非靶向方法侧重于未知代谢物的广泛检测,具有更好的覆盖率,但重现性和定量性能较差,且数据挖掘和代谢物鉴定更复杂,置信水平相对较低[79]. 如Chen等[80]发现,非靶向代谢组学采集的相同谱图数据经不同软件处理后,可能得出不同的数据结论. 靶向和非靶向方法各有优缺点,代谢物高覆盖检测和鉴定,并保持良好的动态范围和定量能力仍然具有挑战性. 代谢组学研究的最新趋势表明,需要将两种主流方法结合起来,形成一种可能具有革命性的方法——混合方法[81].
-
为了在代谢组学分析中同时实现代谢物高覆盖范围和优异的定量性能,出现了将靶向和非靶向方法结合起来的混合方法. 通常混合方法首先对生物样品进行非靶向全局分析或对数据库全面采集来获取特征离子对的信息,再根据生成的离子对信息库进行质谱靶向扫描(MRM、SRM等). 这种方法摆脱了对化学标准品的依赖,同时结合靶向扫描可以在实现高覆盖代谢组的同时表现出高选择性和可靠的定量性能[82].
-
目前已经有许多研究基于单一质谱平台开发了混合方法. 基于QQQ的全局优化靶向质谱(GOT-MS)[83]集成了传统全局分析和靶向检测的优点,首先进行质量范围内选择离子监测(SRM),MS/MS扫描后选择母离子和子离子,在MRM模式下检测,4次进样检测了所有1890个离子对,并在结肠癌的生物标志物发现中验证了方法的高覆盖性和可靠性. GOT-MS基于LC-QQQ即可发现和获取全面的定量MRM,但该方法主要缺点是优化大量MRM所需的时间较多. 目前,GOT-MS被优化开发为数据库辅助的dGOT-MS方法[84],数据库包含了正离子和负离子模式下大量代谢物标准品在多种碰撞能下的质谱信息[14, 85],直接从数据库获取MRM离子对,进行生物样品分析,发现潜在生物标志物. 在乳腺癌相关的生物标志物发现中,dGOT-MS显示有28种潜在生物标志物(FC>1.5且P<0.05),而传统方法只发现5种. 原则上,dGOT-MS能涵盖代谢组学数据库中各种生物样品所表征的所有代谢物. 另外,还有研究人员通过时间交错(ts)/质量交错(ms)方法对GOT-MS进行了优化,通过tsGOT和msGOT对MRM或SRM进行优化,来避免共洗脱并实现对低丰度代谢物的检测[86].
另外也有研究基于单一的高分辨质谱开发混合方法. Gao等[87]基于二维液相色谱/高分辨质谱联用平台开发了STQUM策略,将数据依赖的平行反应监测(PRM)和数据独立的全离子碎裂(AIF)方法相结合,实现了可同步进行靶向定量和非靶向鉴定的高效采集,鉴定和精确定量了88种癌症相关代谢物. 最近,Zhang等[88]基于Orbitrap高分辨质谱开发了结合靶向PRM扫描和非靶向DDA扫描结合的方法,对胰腺肿瘤患者的血浆样本进行分析,证明了该方法对代谢物准确定量和高覆盖分析的能力.
-
除了上述基于单一质谱开发的混合方法,还可以通过多个质谱仪开发结合非靶向和靶向数据采集策略的混合方法. 这种方法也常被称为拟靶向方法,已被认为是第二代代谢组学方法[24]. 拟靶向方法首先基于高分辨质谱采集生物样品的非靶向代谢信息,保证了代谢组的高覆盖性,从非靶向数据中提取物质峰的MS1和二级谱图信息,生成MRM离子对,然后基于三重四极杆进行MRM检测,确保了优异的定量性能. Li等[89]首先基于GC-MS引入了这种方法,通过建立保留时间锁定的GC-MS耦合选择离子监测(RTL-GC/MS-SIM)的方法,将非靶向分析转变为拟靶向方法,对3个不同种植区烟草样品的差异分析证明了方法的更高灵敏度、更好线性和数据质量. Chen等[79]开发了基于LC-MS的拟靶向方法,提供了从真实样品中获取MRM离子对的方法,方法共检测了518种血清代谢物,可用于肝细胞癌生物标志物发现的后续研究. 然而从数千个候选特征中选择离子对是非常耗时的步骤. 为此,开发了一种系统化和自动化的软件“多反应监测-离子对查找器”(MRM-Ion Pair Finder)[90],可以自动化实现母离子比对、MS2提取和还原、特征子离子选择和离子融合的过程,在肝癌代谢组学研究中获得了854个MRM离子对,是拟靶向方法中高通量MRM离子对选择的可靠工具. 另外,Zha等[91]开发了SWATHtoMRM方法,首先基于高分辨质谱进行SWATH扫描采集样品的非靶向数据,通过MS1和MS2质谱峰的峰-峰相关性进行母离子和子离子信息匹配,定义MRM离子对,然后基于低分辨质谱的MRM扫描进行靶向分析,该方法可以实现覆盖率高达1000—2000个代谢物的靶向分析. Chen等[92]开发了数据非依赖的靶向定量代谢组学方法(DITQM),首先在高分辨质谱上采用DIA进行顺序步进靶向MS/MS(sst-MS/MS)采集尽可能多的代谢物MS/MS信息,提取MRM离子对在QQQ上进行MRM扫描,在肺癌代谢组分析中建立了1324种代谢物的MRM分析. 拟靶向方法作为一种新兴方法,已经广泛应用于生物标志物发现[79, 93-96]、疾病的诊治[97-99]、植物样品代谢组学[100-102]、药物效用评价[103, 104]、环境污染物暴露[105, 106]及脂质组学领域[107-109],显示出较好的性能,同时也有许多研究在该方法基础上进行进一步改进[24, 82, 110, 111].
混合方法结合了靶向和非靶向方法的优势,同时弥补了各自的一些缺点,可以实现兼具高覆盖度和高性能的定量分析,正成为高覆盖代谢组学领域的一大发展方向. 在全局代谢组学分析以及生物标志物、疾病诊断等领域具有巨大潜力. 当然混合方法也有其局限性所在,样品运行、方法优化、数据筛选等过程需要相对更长的时间,这是该方法的技术瓶颈. 总的来说,混合方法仍将在高覆盖代谢组学领域不断发展和改进,进一步提升效率和准确性.
-
环境污染物经外暴露进入人体后会参与人体代谢过程,引起代谢系统紊乱并对人体健康带来风险. 这些变化会反映在生物流体或组织代谢物中,因此可以将代谢组学方法应用于环境污染物毒性效应评价的研究中,尤其是高覆盖代谢组学可以对毒性代谢反应进行更全面和准确的评估. 目前,代谢组学在环境毒理学领域具有广泛的应用.
-
工业的发展使重金属成为危险的环境污染物之一. 常见的有毒重金属主要包括砷(As)、镉(Cd)和铅(Pb)等[112].
当前研究发现砷暴露会增加高血压、2型糖尿病和慢性肾病等疾病风险[113-115],但其影响机制尚未完全清楚,代谢组学研究为揭示砷分子作用机制提供了新思路. Wang等[116]采用基于超高效液相色谱/质谱(UHPLC/MS)的非靶向代谢组学方法分析慢性砷暴露引起的大鼠血清代谢组变化,结果发现了18种差异代谢物,表明砷暴露后主要导致氨基酸代谢异常以及溶血脂、鞘脂等脂质代谢异常,该结果有助于进一步探究慢性低水平砷暴露对人体的影响. Li等[117]运用UPLC/Q-TOF平台结合多变量统计方法的非靶向代谢组学调查暴露于相对低水平砷的孕妇体内代谢变化,发现了9种与体内氧化应激和肝脏、肾脏代谢紊乱相关的潜在生物标志物,这些物质可能会对孕妇产生不良影响. Spratlen等[118]运用代谢组学方法研究砷暴露与糖尿病风险增加之间的关联机制,结果发现一碳代谢(OCM)的相关代谢途径可以部分解释砷与糖尿病风险间的关联.
尿液代谢组学是研究镉暴露引起肾毒性机制的一种方法. Chen等[119]对慢性镉暴露的大鼠尿液代谢组进行分析,通过基于质谱的非靶向代谢组方法发现镉会对能量和脂质代谢产生干扰,并诱导氧化应激导致肝肾损伤. Zeng等[120]采用基于质谱的DDA采集结合PRM采集的混合方法进行镉暴露下的人尿液代谢组分析,结果显示肌酸代谢、色氨酸代谢和嘌呤代谢等代谢途径受到影响,这些代谢变化可能与镉应激下的肾毒性有关.
铅(Pb)被广泛用于电池、油漆、管道等工业中,铅暴露会引起诸多不良健康风险,如生殖异常、神经和行为障碍等[121-122]. Eguchi等[123]分别采集了废铅酸电池回收厂附近居民和对照区居民的血液和尿液样品,通过对297个选定的代谢物离子对进行质谱分析研究尿液代谢组,发现了与铅暴露相关的10种候选生物标志物,这些代谢物可能与肾脏中小分子运输中断、血红素生物合成途径和大脑毒性有关,表明了铅可能通过改变这些生物途径对个体健康产生影响. 此外,有研究通过高覆盖代谢组非靶向分析方法对铅暴露男性的血浆样品进行代谢组分析,结果表明铅暴露主要通过诱导氧化应激及对神经和免疫功能损害产生不良健康影响[124].
-
代谢组学技术也被广泛应用于有机污染物毒性效应机制研究中. Sun等[125]基于UHPLC-QTOF仪器进行数据依赖型采集,对苯暴露小鼠的盲肠内容物进行代谢组分析,发现苯暴露会导致小鼠肠道菌群失调和代谢紊乱,并显着影响了类固醇生物合成等代谢途径. 双酚A(BPA)是一种广泛使用的工业化合物,也是一种环境内分泌干扰物,Ji等[126]利用高分辨质谱代谢组学分析方法研究BPA对小鼠肝脏代谢功能的影响,结合生物信息学技术,阐明了BPA影响肝脏代谢功能的作用机制. 近年来,随着微塑料在生态系统中的大量检出,微塑料污染已成为广泛关注的全球环境问题[127],由于粒径小,微塑料很容易进入海洋生物体内并积聚,Huang等[128]通过基于质谱的高覆盖代谢组技术探究聚苯乙烯微塑料对海洋贻贝的毒性作用机制,使用环境浓度对采集的贻贝进行微塑料暴露,结果发现聚苯乙烯微塑料会引起贻贝血淋巴中酪氨酸、苯丙氨酸和维生素B6等5种代谢物的显著变化,并影响抗氧化和免疫系统,但在去除污染物后观察到代谢水平的恢复,表明这种毒性作用可能是可逆的.
另外,持久性有机污染物(POPs)因其持久性、半挥发性、生物累积性和高毒性引起了广泛关注,利用代谢组学方法对POPs类物质进行毒性效应评估可以进一步认识其对环境和人体健康的影响. 短链氯化石蜡已于2017年被列入新的POPs类物质,受到越来越多关注,最近有研究用基于LC/MS的拟靶向代谢组学方法对短链氯化石蜡暴露的大鼠血浆进行代谢组学分析,分别研究了低、中、高三种剂量下的毒性作用和相关代谢反应途径,揭示了121种潜在的毒性生物标志物[129]. 多环芳烃(PAHs)也是典型的POPs类物质,Wang等[130]利用基于LC-MS的代谢组学方法对焦化工污染区和未污染区的儿童和老年人尿液进行分析,研究了易感人群在长期环境PAHs暴露下的代谢变化并识别了潜在生物标志物,研究发现长期暴露于低水平的PAHs会导致人体产生氧化应激相关的代谢变化. 全氟辛酸(PFOA)常用在纺织品、消防泡沫等工业生产中,因其持久性、生物累积性和远距离迁移性已被列入POPs清单中,已有研究发现PFOA会对人类淋巴细胞功能产生影响[131],但毒性机制尚不明确,Li等[132]对健康成年人的淋巴细胞进行PFOA暴露,通过高分辨质谱采集高覆盖代谢物谱图,并用KEGG和HMDB数据库进行代谢组分析,结果发现PFOA干扰了氨基酸、脂质和碳水化合物的代谢,这些代谢变化与淋巴细胞和免疫系统密切相关.
-
由于生物体暴露于多种外源性物质,也可以基于代谢组学方法对联合毒性效应进行评估. 有研究用代谢组学方法分析毒死蜱(有机磷杀虫剂)和镉对大鼠肝脏的联合毒性效应,结果发现镉可以加速肝脏中毒死蜱的降解,两者间有潜在的拮抗作用[133]. Liu等[134]招募志愿者进行户外空气颗粒物低暴露和高暴露实验,通过基于UPLC/Q-Exactive平台的非靶向代谢组学方法对不同浓度重金属暴露环境下的血清代谢物变化进行分析,发现鞘脂代谢途径与重金属介导的心血管相关风险有关,其中Ni和Cd显示出最大的影响. Wang等[105]运用拟靶向代谢组学技术对同时暴露于多环芳烃(PAHs)和短链氯化石蜡(SCCPs)的人肝癌细胞HepG2进行代谢组分析,评估两者的联合毒性作用,发现PAHs暴露主要与三羧酸(TCA)循环和嘌呤代谢紊乱相关,SCCPs暴露会影响甘油磷脂代谢、TCA循环和亚油酸代谢途径,而两者的联合暴露会导致脂肪酸代谢和磷脂代谢的协同上调,TCA循环和糖酵解的协同下调,并对嘌呤代谢产生拮抗作用.
-
代谢组学研究的范围和特点决定了其对高覆盖率的要求,而代谢组学的复杂性又为高覆盖分析带来了阻碍. 目前从代谢物提取、仪器发展到数据处理各个方面的提升加快了高覆盖代谢组学的实现,这个过程中仪器数据采集策略很大程度上决定了代谢谱图信息的准确性和覆盖率,进而影响最终的代谢物鉴定率. 本文对基于质谱的高覆盖代谢组学三大分析方法的数据采集策略的进展进行综述:靶向代谢组学中,将MRM和PRM两种采集策略结合是一种新发展,可以用于较大规模靶向代谢组定量分析. 非靶向代谢组学中,已经开发了许多改进传统DDA和DIA的新策略来提高覆盖率并改善谱图质量. 近年来结合了非靶向和靶向方法优势的混合方法成为代谢组学领域新一代方法,本文分别综述了在单一和多个质谱平台上混合方法的开发,这种方法对实现代谢物高覆盖鉴定并保持良好定量性能的目标具有重大意义,但样品运行和数据筛选更为复杂,在多个仪器间的方法转换和参数优化的要求也更高,这种方法仍将不断被改进优化以更好地应用于代谢组学. 高覆盖代谢组学的发展为外暴露引起的生物毒性效应机制研究提供了方法手段,本文对其在环境毒理学领域的应用进行了综述,包括在重金属、有机污染物和联合毒性效应机制方面的应用进展.
LC-MS的技术提升无疑给高覆盖代谢物可靠检测带来了重大发展. 然而目前高覆盖代谢组学分析仍存在一些有待提高的方面,当前代谢组学提升的覆盖率仍然无法涵盖超痕量代谢物,大规模代谢物的准确分析仍然需要不断提升,且对于高通量样品分析需要更快速且准确性高的方法. 另外需要加强分析方法的标准化,从而有助于不同实验室和仪器间的比对,方法和数据的开放性也对进一步发展有重要推动作用. 在数据采集策略方面,这些问题的解决可以通过在质谱数据采集的速率、灵敏度、动态范围和准确性之间进行权衡,针对不同分析需求进行偏向性改进. 另外,机器学习的方法为采集策略的优化和后续的数据处理提供了新的思路.
基于质谱的高覆盖代谢组学数据采集策略研究进展及在环境毒理学的应用
Research progress of data acquisition strategies for mass spectrometry-based high-coverage metabolomics and its application in environmental toxicology
-
摘要: 代谢组学是分析细胞或生物体中小分子化合物的一门组学科学. 代谢组学的目标是尽可能分析特定生物样品中的所有代谢物,实现高覆盖分析. 基于质谱的技术促进了组学规模的代谢物检测,成为代谢组学的重要分析平台. 质谱数据采集策略的选择对获得全面且高质量的代谢谱图信息至关重要,影响数据采集率进而影响代谢组学的覆盖率. 本文全面综述了当前基于质谱的高覆盖代谢组学数据采集策略的研究进展,总结方法的优势和局限,并概述当前代谢组学方法在环境毒理学领域的新应用,最后讨论当前局限并对未来发展做出展望.Abstract: Metabolomics is an omics science that analyzes small molecule compounds in cells or organisms. The main goal of metabolomics is to analyze all metabolites in the specific biological sample to achieve high coverage analysis. Mass spectrometry-based technologies promotes omics-scale metabolite detection and have become the important analytical platforms for metabolomics. The choice of mass spectrometry data acquisition strategy is critical for obtaining comprehensive and high-quality metabolic profile information, which affects data acquisition rates and metabolomics coverage. This paper comprehensively reviewed the current research progress of data collection strategies for mass spectrometry-based high-coverage metabolomics, summarized the advantages and disadvantages of methods, introduced the new applications of metabolomics methods in the field of environmental toxicology, and discussed the current limitations and makes perspectives for the future.
-
代谢组这一术语于1998年提出[1],是细胞或生物体中参与代谢反应的小分子化合物的集合. 类似于基因组学、转录组学和蛋白质组学,代谢组学是系统生物学的一门“组学”科学,旨在对生物体中存在的内源性和外源性代谢物进行分析[2]. 代谢组学的研究对象涵盖了各种不同极性的化合物,这些化合物具有广泛的化学类别,且浓度变化范围达到9个数量级[3],因此如何提升代谢物分析方法的覆盖率以更全面的识别代谢物是代谢组学的首要挑战之一. 高覆盖代谢组学可以同时进行大量代谢物的定性定量分析,并探究与生物表型的因果相关性,识别潜在的生物标志物[4-5],因此高覆盖代谢组学也是评估外暴露引起的生物毒性效应的重要方法,将高覆盖代谢组学方法用于环境毒理学中可以识别污染物暴露引起的代谢物变化,研究污染物与表型之间的毒性效应机制.
高覆盖代谢组学分析的主要流程包括样品前处理,仪器分析和数据分析. 目前已经有许多研究针对代谢物提取方法进行开发与优化[6-10]. 另外随着技术进步,仪器分析平台也更多样化,包括分离技术平台(例如液相色谱(LC)、气相色谱(GC)和毛细管电泳(CE)等)和检测技术平台(例如质谱(MS)和核磁共振(NMR)). 其中液相色谱-质谱联用(LC-MS)仪器在引入电喷雾电离(ESI)后提高了灵敏度,促进了组学规模的代谢物检测[11-12],已经成为代谢组学的重要仪器分析平台. 在数据分析方面,已经开发了更全面的代谢物数据库[13-14],助力代谢物的识别与发现. 计算机的最新技术和生物信息学工具飞速发展,提升了代谢组学数据的处理速度,并将继续推动代谢组学领域的发展. 然而,先进的数据处理工具也无法分析未被仪器采集到的代谢物数据,因此,仪器分析方法的选择对代谢物数据的全面采集和后续的数据分析是十分重要的.
LC-MS平台在进行仪器分析时,代谢物首先以不同的保留时间(RT)经LC洗脱后,进入MS采集母离子的一级谱图(MS1),之后对所选择的母离子进行碎裂采集二级碎片谱图(MS/MS),二级碎片谱图是代谢物结构鉴定的关键. 因此,在基于质谱的仪器分析方法中,数据采集策略决定了如何选择母离子以进一步裂解,是影响用于后续分析的MS/MS的覆盖范围和质量的重要因素.
现有的相关研究多是对基于质谱的代谢组学分析方法的全过程概述,包括了从色谱分离方法到质谱采集以及数据处理的全过程[15-16],但当前还没有聚焦于质谱数据采集策略研究进展的最新综述,本文将补充此方向的研究空白,重点综述2015年以来基于LC-MS的高覆盖代谢组学数据采集策略的研究进展. 当前应用于代谢组学的质谱分析方法包括靶向、非靶向以及两者结合的混合方法,本文将综述针对这3种方法开发的数据采集策略的进展,并介绍高覆盖代谢组学在环境毒理学领域的最新应用.
1. 高覆盖靶向代谢组学数据采集策略(Data acquisition strategies for high-coverage targeted metabolomics)
靶向代谢组学方法是对已知代谢物进行定性和定量的方法,当前靶向代谢组学常用的数据采集策略主要包括多反应监测(MRM)和平行反应监测(PRM).
1.1 多反应监测
基于三重四极杆质谱(QQQ)的多反应监测(MRM)是靶向代谢组学中应用最广泛的数据采集策略,被称为小分子和代谢物定量的金标准[17-18]. 在用MRM进行代谢物分析前,需要先通过标准品对目标代谢物的质谱参数(碰撞能和去簇电压)和离子对信息(前体-产物离子对)进行优化,选择最优灵敏度. 在MRM采集时,Q1选择感兴趣的目标母离子,Q2作为碰撞池对目标前体进行碎裂,Q3对预选的子离子进行分离(方法流程图可见下图1). 虽然方法建立之初需要对每个目标物进行参数优化,但该方法定量灵敏度和准确性很高,可以对极少量的样品或极低浓度的样品进行准确的定量分析,如有研究用10 μL全血对36种目标代谢物进行靶向分析,结果呈现优异的稳定性和可重复性[19]. 然而由于依赖于标准品,这种传统MRM方法的代谢物覆盖范围有限.
 图 1 分别在QQQ和Q-Orbitrap仪器上进行MRM(a)和PRM(b)的示意图Figure 1. Schematic representation of MRM (a) and PRM (b) as performed on QQQ and Q-Orbitrap instrumentation respectively
图 1 分别在QQQ和Q-Orbitrap仪器上进行MRM(a)和PRM(b)的示意图Figure 1. Schematic representation of MRM (a) and PRM (b) as performed on QQQ and Q-Orbitrap instrumentation respectively目前,已经开发了新的方法提升基于MRM的靶向代谢组学覆盖率. 基于合成大量标准品进行靶向分析的靶向试剂盒技术增加了目标代谢物的覆盖范围,该方法使基于MRM的靶向方法可以实现快速定量数百种目标代谢物,例如有研究使用AbsoluteIDQ™ p180 kit代谢组试剂盒进行靶向代谢组分析,可靠鉴定了159种血清代谢物[20];而一些靶向代谢组学试剂盒能对500多种化合物进行检测量化[21-22]. 虽然方法得到了很好的优化,但这种多反应监测方法仍然依赖于目标物的标准品. 为了摆脱对标准品的依赖,进一步增加多反应监测方法的覆盖度,Domingo-Almenara等[23]建立了用于小分子靶向分析的MRM离子对库,该数据库包含了超过15500个分子的实验或计算机生成MRM离子对,可以实现大量内源性和外源性代谢物的靶向识别与量化. 这种基于建立的MRM数据库进行样品MRM采集的方法可以分析没有标准品的物质,提高分析覆盖度,但不同仪器或样本类型之间的兼容性通常不好[24].
1.2 平行反应监测
高分辨率质谱仪(HRMS)的发展为化合物分析提供了新方法. 基于HRMS进行的靶向数据采集,常称为平行反应监测(PRM),也被描述为高分辨MRM[25]. 其工作流程如图1所示,与MRM类似,PRM采集模式同样是在Q1进行母离子的筛选,在Q2进行母离子碎裂,不同之处在于,PRM第三步通过高分辨的质谱进行所有碎片离子的测定.
基于HRMS的PRM采集模式能以17500—35000分辨率进行MS分析,可以分辨小于0.01 Da的质量差异,从而可以有效地将目标物与其他干扰离子区分开[25],因此可以采集高准确度MS/MS,识别目标代谢物的离子进行定量,对标准品的依赖较低,这是PRM的一大优势. PRM已经应用于多个生物学领域的靶向肽定量[26-27]. Zhou等[25]在Q-Orbitrap仪器上开发了针对237种极性代谢物的PRM方法,通过在不同种生物样品中进行应用,证明了PRM在大规模靶向代谢物定量方面出色的灵敏度、准确性和适用性. 另外,PRM由于监测了所有二级碎片离子,优化过程更简单,相比MRM在方法开发阶段耗时少. 但鉴于高分辨质谱扫描速度较慢,PRM在同时进行大规模靶向分析方面存在限制. 相较而言,QQQ仪器具有高扫描速度和灵敏度,这缩短了MRM采集的循环时间,有利于更高效的分析,并确保足够的色谱峰数据点进行准确定量[25].
MRM方法优势在于高扫描速度和极性切换,而PRM具有高分辨率和全碎片采集的优点. 在大规模靶向代谢组定量分析时,可以考虑结合两者的优势,将PRM作为MRM的补充工具,先经PRM获得代谢物二级谱图信息,再转为MRM进行高速扫描分析[25].
2. 高覆盖非靶向代谢组学数据采集策略(Data acquisition strategies for high-coverage untargeted metabolomics)
非靶向代谢组学是在没有标准品的情况下无偏向性的对所有采集到的谱图进行检测,以达到高覆盖的非靶向代谢组学分析. 非靶向代谢物鉴定通常依赖于高分辨MS1谱图和MS/MS谱图,其中MS1谱图用于确定分子式,MS/MS谱图用于代谢物结构鉴定,因此首先需要采集全面且准确的代谢物MS1和MS/MS谱图信息. 高分辨率质谱已被证明是非靶向代谢组学分析的强大信息采集工具,因其能获得高分辨率和高质量准确度的扫描信息. 基于高分辨质谱的非靶向代谢组学获取谱图数据主要有两种方式:对用于全局分析的MS1数据和用于结构鉴定的MS/MS数据分别进行采集;或者在一次运行中同时采集MS1和MS/MS数据[28].
2.1 全扫描
全扫描(Full scan)是对MS1和MS/MS数据分别进行采集的方法,是代谢组学中常用到的数据采集方法[29-30]. 首先对样品进行全扫描,通过较低的碰撞能量即可获得所有代谢特征的MS1信息. 然后经预处理软件(如XCMS等)提取代谢特征[31],基于给定生物学问题选择感兴趣的特征在后续实验中进行MS/MS采集,鉴定代谢物. 全扫描在采集、数据处理方面比较简单,但它将代谢物定量与结构鉴定分离开来,需要额外的仪器分析时间和样品量[32],并且多次分析之间间隔的时间可能会出现数据难以对齐和样品变化等问题[28].
近年来越来越受到关注的另一种方式是在一次运行中同时采集MS1和MS/MS数据. 这种方法主要有两种模式:数据依赖型扫描(data dependent acquisition,DDA)和数据非依赖型扫描(data independent acquisition,DIA).
2.2 数据依赖型扫描
2.2.1 传统的数据依赖型扫描方法
DDA是在全扫描基础上开发的一种更省时快速的数据采集模式,是目前非靶向分析中常用的方法. DDA模式下,质谱仪自动地在Full scan MS和MS/MS采集间切换,在每个循环中先进行MS全扫描采集一级质谱信息,然后选择目标母离子进行碎裂并获得二级谱图,进行物质鉴定,如图2所示[33]. 目标母离子的筛选标准有离子丰度,信噪比,同位素分布等,最常见的是选择强度最高的前几个离子进行碎裂. DDA在一次分析中同时获得MS1和筛选母离子的MS/MS[32],可以同时进行数据处理和代谢物鉴定. 且DDA采用较窄的m/z窗口筛选目标母离子,减少了干扰离子的存在,可以提供较高质量的MS/MS. 但由于目标母离子的筛选通常取决于强度,一些感兴趣的低丰度母离子不能被碎裂,无法获取二级谱图,导致传统DDA方法难以鉴定低丰度物质,且由于筛选过程存在一定随机性,在每次分析时选取的母离子并不完全相同,导致方法重现性较低[34].
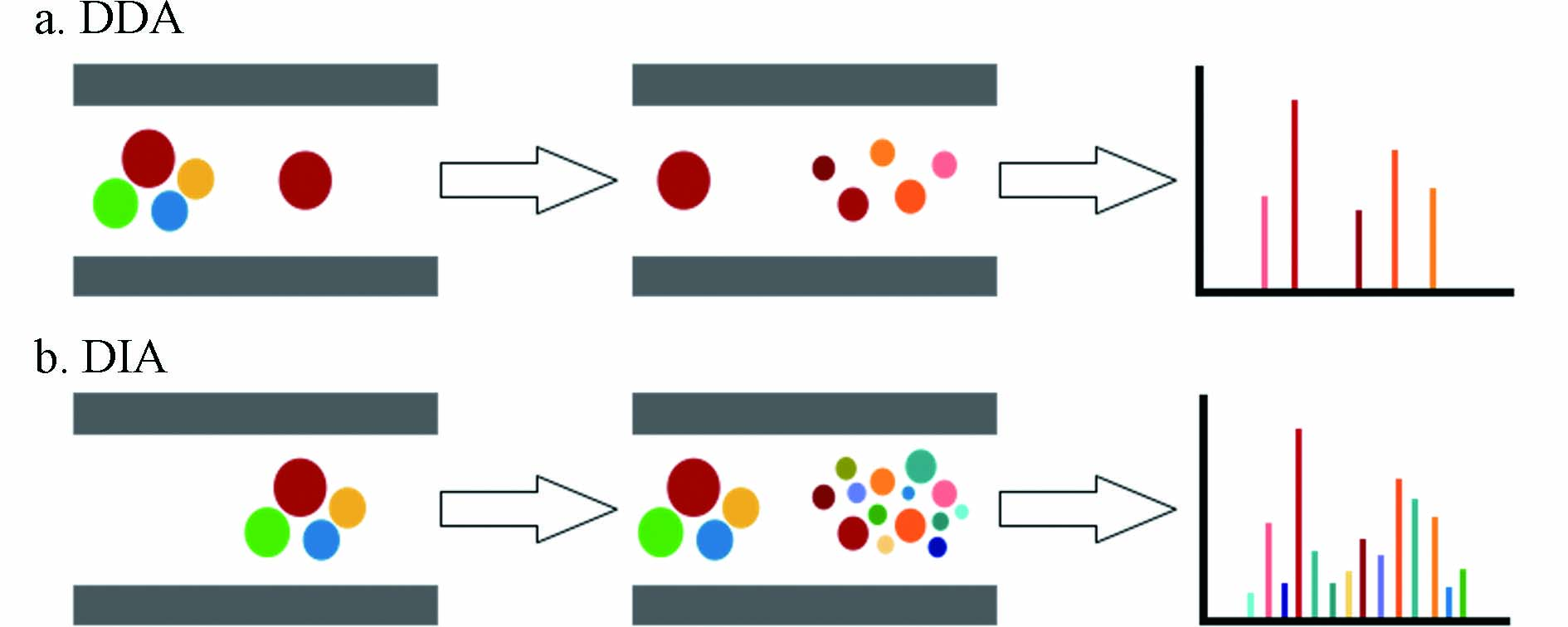 图 2 两种非靶向采集模式的工作流程及结果示意图Figure 2. Schematic representation of the workflows and results obtained from two non-target acquisition modes
图 2 两种非靶向采集模式的工作流程及结果示意图Figure 2. Schematic representation of the workflows and results obtained from two non-target acquisition modes2.2.2 数据依赖型扫描方法的改进
(1)基于排除或包含列表的数据依赖型扫描方法
由于传统的DDA方法难以鉴定低丰度物质,已有研究通过对传统DDA方法的改进,提高了方法的覆盖范围和性能. 首先是基于DDA设置排除列表的方法,将背景离子或不需碎裂的离子列入排除列表,在下一次仪器分析时不进行碎裂. 静态排除列表需要首先运行空白样品,以生成背景离子排除列表,这种方法通过过滤无关特征来增加代谢物的二级谱图覆盖率,可以排除基线噪声或背景干扰,但对鬼峰的排除效率较低[35]. 近年来结合多针进样的动态排除列表方法得到发展,这种方法在第一针采用DDA方法进样,将获得的有二级谱图的母离子及其保留时间范围输入排除列表中,使得已采集二级谱图的母离子在下一针进样时不再被选择,从而使更低丰度的母离子可以被选择[35],通过多针进样提高二级谱图覆盖率. 这种方法最早应用于蛋白质组学,Bendall等[36]利用这一原理开发了迭代排除质谱法(IE-MS),与5次重复DDA相比,5次IE-MS识别的独特蛋白质增加了30%,降低了对低丰度肽的数据采集偏差. 动态列表方法也被应用于代谢组学中,Edmands等[37]通过6次重复进样的迭代排除方法对经常食用富含多酚食物的人的尿液进行分析,确定了与所选食物摄入相关的80多种多酚代谢物. 由于手动生成排除列表的工作耗时,Koelmel等[38]开发了R脚本“IE-omics”,输入原始数据即可自动生成排除列表,将其应用于脂质组学可将识别率提高50%以上. 理论上,迭代排除DDA方法可通过不断迭代来获取所有高于背景信号的母离子的二级谱图,但在分析大批量样品时要评估重复进样增加的仪器时间成本. 有研究发现在二级谱图质量和覆盖率之间的最好折衷方案是进行五轮迭代排除,这样即可大大提高二级覆盖率[39],为鉴定提供大量信息.
基于DDA设置包含列表的方法,是通过将样品相关离子列入包含列表,优先选择包含列表中母离子进行碎裂,以增加特异性MS/MS覆盖率[40]. Roger Giné等[41]介绍了一种以分子式为导向的采集方法HERMES,根据分析样品的特异性信息选择独特分子式,在进行二级谱图扫描时选择包含列表中的这些样品相关前体离子,从而提高代谢物分析的选择性和二级谱图的覆盖度,结果表明HERMES鉴定的代谢物是三轮迭代排除DDA方法的两倍.
目前也有研究将包含列表与排除列表相结合进一步增加二级谱图覆盖范围. Cho等[28]基于空白扣除过滤非生物特征,基于实际样品分析生成包含独特生物代谢物的离子列表,集成了排除列表和包含列表,这种方法在四次进样后获取了>99%的独特代谢物的二级谱图,显著提高了覆盖率. 近年来也提出了更加自动化的手段推动了方法的应用. AcquireX智能化数据采集模式[42]基于机器学习的方法自动迭代更新背景排除和前体包含列表,在同一样品重复进样中,可以实现相关特征的高覆盖DDA采集.
(2)数据依赖型扫描方法的智能化改进
除了通过排除和包含列表进行DDA方法的改进,目前还开发了其他智能化手段进行DDA方法的改进. Broeckling等[43]考虑到代谢组学数据采集建立在大队列的样品组上,提出了一种数据集依赖型采集模式(DsDA),通过实时分析已采集数据,及时优化后续序列中MS/MS采集方案,并利用R等软件实现了自动化数据分析. 通过在数据处理和数据采集之间的近乎实时反馈循环,可以获得全面的二级覆盖范围. 最近,Bills等[44]建立了一种新的采集策略,称为Met-IQ,将实验MS/MS实时地与参考数据库比较,谱图相似则自动执行多级质谱(MSn)采集,获得更多结构信息,通过将MS3采集限制在可能的目标化合物上,从而减少了MS3的触发,提高了MS2采集率. Wandy等[45]介绍了一种虚拟代谢组学质谱仪(ViMMS),是一种用于代谢组学的模块化LC-MS/MS模拟器,可以对不同策略进行测试并获得模拟结果,允许在计算机上对二级采集进行优化而无需占用仪器时间. Davies等[46]扩展了ViMMS的使用,通过仪器应用程序编程接口(IAPI)创建了允许ViMMS开发的控制器直接在实际质谱上使用的桥接代码. 用这种方法开发了两种新的DDA策略并应用于实际代谢物分析,结果均优于传统DDA的覆盖范围. 这种基于计算机模拟、优化和验证的过程为未来的采集策略改进提供了方向.
这些方法的改进在不同程度上提高了DDA的二级覆盖率,并通过与可用数据库的匹配进行鉴定. 如Chen等[47]在Q-TOF仪器上开发了一种DDA方法研究人类泪液代谢组,将MS/MS谱图与METLIN[48]、HMDB[14]和Massbank[49]数据库进行匹配,鉴定出60种代谢物. 所有这些研究为集成HRMS和DDA工作流程以识别、鉴定和量化生物提取物中的代谢特征铺平了道路. 然而,为了更好地弥补由于母离子选择和欠采样带来的限制,可以无差别对每个母离子进行碎裂的数据非依赖型采集(DIA)工作流程逐渐被开发和广泛应用[34].
2.3 数据非依赖型扫描
2.3.1 传统的数据非依赖型扫描方法
数据非依赖型扫描(data independent acquisition,DIA)下,质谱仪通过高低碰撞能量交替采集信息,低能量时采集一级质谱,高能量时打碎离子无偏获取全部二级碎片信息(图2分别展示了DDA和DIA的工作流程). 传统的DIA方法包括:MSE、MSALL或MS-AIF[35, 50]. Wrona等[51]首先在LC-QTOF上开发了这种数据采集方法,并成功应用于维拉帕米(心血管药物)体外代谢物的鉴定. 之后Plumb等[52]描述了UPLC/MSE的方法,通过高低碰撞能交替获得所有离子MS/MS数据,并在大鼠尿液的内源物分析中验证了该方法. 类似的全离子碎裂(AIF)方法也已经在Orbitrap仪器上开发并应用[53-56]. 与DDA不同,DIA不预先筛选母离子,理论上能获取所有母离子的碎片信息. 然而明显的缺陷在于母离子与其碎片离子之间的联系被解离,所得碎片谱图复杂而无法直接将碎片对应到特定的母离子[57],对后续代谢物的鉴定带来了挑战. 因此需要对传统DIA方法进行改进,同时结合谱图解卷积的软件来应对这一挑战.
2.3.2 数据非依赖型扫描方法的改进
当前已经开发了多种方法对传统DIA进行改进,例如将离子淌度分析仪(IMS)耦合到DIA工作流程中. IMS通过额外确定碰撞截面(CCS)值(与离子形状相关的参数),可以提高代谢物鉴定的准确性,甚至解决同分异构体的分离问题[58]. Paglia等[58]将离子淌度集成到DIA中,可以分离共流出的L-磷脂酰乙醇胺,证明了在碎片化前进行离子淌度分离能提供更清晰的MS/MS谱图,促进建立母离子和碎片离子之间的联系,从而提高鉴定的特异性. 同时IMS获得的CCS值具有非常高的重现性(RSD<2%)[59].
为了提高特异性,Gillet等[60]引入了另一种改进方法,即基于Q-TOF的所有理论质谱的顺序窗口采集(SWATH),该方法将扫描范围划分为以某一固定宽度(如20或25Da)为间隔的连续区间. Zhang等[61]通过在SWATH采集中使用可变隔离窗口进一步提高了LC-SWATH-MS采集的选择性. 目前SWATH方法已广泛应用于非靶向代谢组学[57, 62-67]. 近年来还提出了其他改进策略进一步提高选择性. Moseley等[68]描述了一种称为SONAR的新型扫描四极杆DIA方法,对质量范围内的母离子进行快速移动的四极杆扫描,根据四极杆移动位置将母离子与子离子相关联,具有更高的选择性. 另有研究开发了改进的ScanningSWATH[69],将SWATH的Q1分段式质量窗口采集转变为按一定窗口大小连续的Q1采集,通过更小的隔离窗口提供更好的谱图专属性. 此外还有通过小隔离窗口获得MS/MS谱图的PASS-DIA方法[70]. 最近有研究提出了一种数据依赖辅助数据独立采集(DaDIA)的新工作流程,对单个样品执行DIA,合并样品进行DDA,结合了DIA的高MS2覆盖率和DDA的高MS2谱图质量[71]. 除上述方法外,还有一些更早的DIA改进策略,可以参见Chapman等于2014年发表的综述[72].
尽管如此,这些改进的DIA技术仍不能完全可靠地建立母离子和碎片离子之间的联系,需要使用算法软件对嵌合的MS/MS谱图解卷积. 目前已成功开发了一些DIA数据的解卷积软件,如CorrDec、MS-DIAL、DecoMetDIA或MetDIA等[73-76]. 解卷积后的谱图可以与谱库比对鉴定,如今质谱数据库不断扩大,如MassBank[49]、METLIN[77]、HMDB[14]或GNPS[78]等,但数量和可用性仍不够,且扩展这些实验衍生的质谱数据库非常耗时,当前在开发谱图预测和代谢物注释的计算机生成质谱的计算工具方面付出了相当大的努力.
总的来说,靶向方法旨在识别和量化有限数量的已知代谢物,具有可靠的定量准确性、宽动态范围和高灵敏度,但通常无法检测未知物. 非靶向方法侧重于未知代谢物的广泛检测,具有更好的覆盖率,但重现性和定量性能较差,且数据挖掘和代谢物鉴定更复杂,置信水平相对较低[79]. 如Chen等[80]发现,非靶向代谢组学采集的相同谱图数据经不同软件处理后,可能得出不同的数据结论. 靶向和非靶向方法各有优缺点,代谢物高覆盖检测和鉴定,并保持良好的动态范围和定量能力仍然具有挑战性. 代谢组学研究的最新趋势表明,需要将两种主流方法结合起来,形成一种可能具有革命性的方法——混合方法[81].
3. 代谢组学混合方法的数据采集策略(Data acquisition strategies for hybrid metabolomics approaches)
为了在代谢组学分析中同时实现代谢物高覆盖范围和优异的定量性能,出现了将靶向和非靶向方法结合起来的混合方法. 通常混合方法首先对生物样品进行非靶向全局分析或对数据库全面采集来获取特征离子对的信息,再根据生成的离子对信息库进行质谱靶向扫描(MRM、SRM等). 这种方法摆脱了对化学标准品的依赖,同时结合靶向扫描可以在实现高覆盖代谢组的同时表现出高选择性和可靠的定量性能[82].
3.1 基于单一质谱平台开发的混合方法数据采集策略
目前已经有许多研究基于单一质谱平台开发了混合方法. 基于QQQ的全局优化靶向质谱(GOT-MS)[83]集成了传统全局分析和靶向检测的优点,首先进行质量范围内选择离子监测(SRM),MS/MS扫描后选择母离子和子离子,在MRM模式下检测,4次进样检测了所有1890个离子对,并在结肠癌的生物标志物发现中验证了方法的高覆盖性和可靠性. GOT-MS基于LC-QQQ即可发现和获取全面的定量MRM,但该方法主要缺点是优化大量MRM所需的时间较多. 目前,GOT-MS被优化开发为数据库辅助的dGOT-MS方法[84],数据库包含了正离子和负离子模式下大量代谢物标准品在多种碰撞能下的质谱信息[14, 85],直接从数据库获取MRM离子对,进行生物样品分析,发现潜在生物标志物. 在乳腺癌相关的生物标志物发现中,dGOT-MS显示有28种潜在生物标志物(FC>1.5且P<0.05),而传统方法只发现5种. 原则上,dGOT-MS能涵盖代谢组学数据库中各种生物样品所表征的所有代谢物. 另外,还有研究人员通过时间交错(ts)/质量交错(ms)方法对GOT-MS进行了优化,通过tsGOT和msGOT对MRM或SRM进行优化,来避免共洗脱并实现对低丰度代谢物的检测[86].
另外也有研究基于单一的高分辨质谱开发混合方法. Gao等[87]基于二维液相色谱/高分辨质谱联用平台开发了STQUM策略,将数据依赖的平行反应监测(PRM)和数据独立的全离子碎裂(AIF)方法相结合,实现了可同步进行靶向定量和非靶向鉴定的高效采集,鉴定和精确定量了88种癌症相关代谢物. 最近,Zhang等[88]基于Orbitrap高分辨质谱开发了结合靶向PRM扫描和非靶向DDA扫描结合的方法,对胰腺肿瘤患者的血浆样本进行分析,证明了该方法对代谢物准确定量和高覆盖分析的能力.
3.2 多质谱平台结合开发的混合方法数据采集策略
除了上述基于单一质谱开发的混合方法,还可以通过多个质谱仪开发结合非靶向和靶向数据采集策略的混合方法. 这种方法也常被称为拟靶向方法,已被认为是第二代代谢组学方法[24]. 拟靶向方法首先基于高分辨质谱采集生物样品的非靶向代谢信息,保证了代谢组的高覆盖性,从非靶向数据中提取物质峰的MS1和二级谱图信息,生成MRM离子对,然后基于三重四极杆进行MRM检测,确保了优异的定量性能. Li等[89]首先基于GC-MS引入了这种方法,通过建立保留时间锁定的GC-MS耦合选择离子监测(RTL-GC/MS-SIM)的方法,将非靶向分析转变为拟靶向方法,对3个不同种植区烟草样品的差异分析证明了方法的更高灵敏度、更好线性和数据质量. Chen等[79]开发了基于LC-MS的拟靶向方法,提供了从真实样品中获取MRM离子对的方法,方法共检测了518种血清代谢物,可用于肝细胞癌生物标志物发现的后续研究. 然而从数千个候选特征中选择离子对是非常耗时的步骤. 为此,开发了一种系统化和自动化的软件“多反应监测-离子对查找器”(MRM-Ion Pair Finder)[90],可以自动化实现母离子比对、MS2提取和还原、特征子离子选择和离子融合的过程,在肝癌代谢组学研究中获得了854个MRM离子对,是拟靶向方法中高通量MRM离子对选择的可靠工具. 另外,Zha等[91]开发了SWATHtoMRM方法,首先基于高分辨质谱进行SWATH扫描采集样品的非靶向数据,通过MS1和MS2质谱峰的峰-峰相关性进行母离子和子离子信息匹配,定义MRM离子对,然后基于低分辨质谱的MRM扫描进行靶向分析,该方法可以实现覆盖率高达1000—2000个代谢物的靶向分析. Chen等[92]开发了数据非依赖的靶向定量代谢组学方法(DITQM),首先在高分辨质谱上采用DIA进行顺序步进靶向MS/MS(sst-MS/MS)采集尽可能多的代谢物MS/MS信息,提取MRM离子对在QQQ上进行MRM扫描,在肺癌代谢组分析中建立了1324种代谢物的MRM分析. 拟靶向方法作为一种新兴方法,已经广泛应用于生物标志物发现[79, 93-96]、疾病的诊治[97-99]、植物样品代谢组学[100-102]、药物效用评价[103, 104]、环境污染物暴露[105, 106]及脂质组学领域[107-109],显示出较好的性能,同时也有许多研究在该方法基础上进行进一步改进[24, 82, 110, 111].
混合方法结合了靶向和非靶向方法的优势,同时弥补了各自的一些缺点,可以实现兼具高覆盖度和高性能的定量分析,正成为高覆盖代谢组学领域的一大发展方向. 在全局代谢组学分析以及生物标志物、疾病诊断等领域具有巨大潜力. 当然混合方法也有其局限性所在,样品运行、方法优化、数据筛选等过程需要相对更长的时间,这是该方法的技术瓶颈. 总的来说,混合方法仍将在高覆盖代谢组学领域不断发展和改进,进一步提升效率和准确性.
4. 在环境毒理学中的应用(Applications in environmental toxicology)
环境污染物经外暴露进入人体后会参与人体代谢过程,引起代谢系统紊乱并对人体健康带来风险. 这些变化会反映在生物流体或组织代谢物中,因此可以将代谢组学方法应用于环境污染物毒性效应评价的研究中,尤其是高覆盖代谢组学可以对毒性代谢反应进行更全面和准确的评估. 目前,代谢组学在环境毒理学领域具有广泛的应用.
4.1 在重金属毒性效应机制研究中的应用
工业的发展使重金属成为危险的环境污染物之一. 常见的有毒重金属主要包括砷(As)、镉(Cd)和铅(Pb)等[112].
当前研究发现砷暴露会增加高血压、2型糖尿病和慢性肾病等疾病风险[113-115],但其影响机制尚未完全清楚,代谢组学研究为揭示砷分子作用机制提供了新思路. Wang等[116]采用基于超高效液相色谱/质谱(UHPLC/MS)的非靶向代谢组学方法分析慢性砷暴露引起的大鼠血清代谢组变化,结果发现了18种差异代谢物,表明砷暴露后主要导致氨基酸代谢异常以及溶血脂、鞘脂等脂质代谢异常,该结果有助于进一步探究慢性低水平砷暴露对人体的影响. Li等[117]运用UPLC/Q-TOF平台结合多变量统计方法的非靶向代谢组学调查暴露于相对低水平砷的孕妇体内代谢变化,发现了9种与体内氧化应激和肝脏、肾脏代谢紊乱相关的潜在生物标志物,这些物质可能会对孕妇产生不良影响. Spratlen等[118]运用代谢组学方法研究砷暴露与糖尿病风险增加之间的关联机制,结果发现一碳代谢(OCM)的相关代谢途径可以部分解释砷与糖尿病风险间的关联.
尿液代谢组学是研究镉暴露引起肾毒性机制的一种方法. Chen等[119]对慢性镉暴露的大鼠尿液代谢组进行分析,通过基于质谱的非靶向代谢组方法发现镉会对能量和脂质代谢产生干扰,并诱导氧化应激导致肝肾损伤. Zeng等[120]采用基于质谱的DDA采集结合PRM采集的混合方法进行镉暴露下的人尿液代谢组分析,结果显示肌酸代谢、色氨酸代谢和嘌呤代谢等代谢途径受到影响,这些代谢变化可能与镉应激下的肾毒性有关.
铅(Pb)被广泛用于电池、油漆、管道等工业中,铅暴露会引起诸多不良健康风险,如生殖异常、神经和行为障碍等[121-122]. Eguchi等[123]分别采集了废铅酸电池回收厂附近居民和对照区居民的血液和尿液样品,通过对297个选定的代谢物离子对进行质谱分析研究尿液代谢组,发现了与铅暴露相关的10种候选生物标志物,这些代谢物可能与肾脏中小分子运输中断、血红素生物合成途径和大脑毒性有关,表明了铅可能通过改变这些生物途径对个体健康产生影响. 此外,有研究通过高覆盖代谢组非靶向分析方法对铅暴露男性的血浆样品进行代谢组分析,结果表明铅暴露主要通过诱导氧化应激及对神经和免疫功能损害产生不良健康影响[124].
4.2 在有机污染物毒性效应机制研究中的应用
代谢组学技术也被广泛应用于有机污染物毒性效应机制研究中. Sun等[125]基于UHPLC-QTOF仪器进行数据依赖型采集,对苯暴露小鼠的盲肠内容物进行代谢组分析,发现苯暴露会导致小鼠肠道菌群失调和代谢紊乱,并显着影响了类固醇生物合成等代谢途径. 双酚A(BPA)是一种广泛使用的工业化合物,也是一种环境内分泌干扰物,Ji等[126]利用高分辨质谱代谢组学分析方法研究BPA对小鼠肝脏代谢功能的影响,结合生物信息学技术,阐明了BPA影响肝脏代谢功能的作用机制. 近年来,随着微塑料在生态系统中的大量检出,微塑料污染已成为广泛关注的全球环境问题[127],由于粒径小,微塑料很容易进入海洋生物体内并积聚,Huang等[128]通过基于质谱的高覆盖代谢组技术探究聚苯乙烯微塑料对海洋贻贝的毒性作用机制,使用环境浓度对采集的贻贝进行微塑料暴露,结果发现聚苯乙烯微塑料会引起贻贝血淋巴中酪氨酸、苯丙氨酸和维生素B6等5种代谢物的显著变化,并影响抗氧化和免疫系统,但在去除污染物后观察到代谢水平的恢复,表明这种毒性作用可能是可逆的.
另外,持久性有机污染物(POPs)因其持久性、半挥发性、生物累积性和高毒性引起了广泛关注,利用代谢组学方法对POPs类物质进行毒性效应评估可以进一步认识其对环境和人体健康的影响. 短链氯化石蜡已于2017年被列入新的POPs类物质,受到越来越多关注,最近有研究用基于LC/MS的拟靶向代谢组学方法对短链氯化石蜡暴露的大鼠血浆进行代谢组学分析,分别研究了低、中、高三种剂量下的毒性作用和相关代谢反应途径,揭示了121种潜在的毒性生物标志物[129]. 多环芳烃(PAHs)也是典型的POPs类物质,Wang等[130]利用基于LC-MS的代谢组学方法对焦化工污染区和未污染区的儿童和老年人尿液进行分析,研究了易感人群在长期环境PAHs暴露下的代谢变化并识别了潜在生物标志物,研究发现长期暴露于低水平的PAHs会导致人体产生氧化应激相关的代谢变化. 全氟辛酸(PFOA)常用在纺织品、消防泡沫等工业生产中,因其持久性、生物累积性和远距离迁移性已被列入POPs清单中,已有研究发现PFOA会对人类淋巴细胞功能产生影响[131],但毒性机制尚不明确,Li等[132]对健康成年人的淋巴细胞进行PFOA暴露,通过高分辨质谱采集高覆盖代谢物谱图,并用KEGG和HMDB数据库进行代谢组分析,结果发现PFOA干扰了氨基酸、脂质和碳水化合物的代谢,这些代谢变化与淋巴细胞和免疫系统密切相关.
4.3 在联合毒性效应机制研究中的应用
由于生物体暴露于多种外源性物质,也可以基于代谢组学方法对联合毒性效应进行评估. 有研究用代谢组学方法分析毒死蜱(有机磷杀虫剂)和镉对大鼠肝脏的联合毒性效应,结果发现镉可以加速肝脏中毒死蜱的降解,两者间有潜在的拮抗作用[133]. Liu等[134]招募志愿者进行户外空气颗粒物低暴露和高暴露实验,通过基于UPLC/Q-Exactive平台的非靶向代谢组学方法对不同浓度重金属暴露环境下的血清代谢物变化进行分析,发现鞘脂代谢途径与重金属介导的心血管相关风险有关,其中Ni和Cd显示出最大的影响. Wang等[105]运用拟靶向代谢组学技术对同时暴露于多环芳烃(PAHs)和短链氯化石蜡(SCCPs)的人肝癌细胞HepG2进行代谢组分析,评估两者的联合毒性作用,发现PAHs暴露主要与三羧酸(TCA)循环和嘌呤代谢紊乱相关,SCCPs暴露会影响甘油磷脂代谢、TCA循环和亚油酸代谢途径,而两者的联合暴露会导致脂肪酸代谢和磷脂代谢的协同上调,TCA循环和糖酵解的协同下调,并对嘌呤代谢产生拮抗作用.
5. 总结与展望(Summaries and prospects)
代谢组学研究的范围和特点决定了其对高覆盖率的要求,而代谢组学的复杂性又为高覆盖分析带来了阻碍. 目前从代谢物提取、仪器发展到数据处理各个方面的提升加快了高覆盖代谢组学的实现,这个过程中仪器数据采集策略很大程度上决定了代谢谱图信息的准确性和覆盖率,进而影响最终的代谢物鉴定率. 本文对基于质谱的高覆盖代谢组学三大分析方法的数据采集策略的进展进行综述:靶向代谢组学中,将MRM和PRM两种采集策略结合是一种新发展,可以用于较大规模靶向代谢组定量分析. 非靶向代谢组学中,已经开发了许多改进传统DDA和DIA的新策略来提高覆盖率并改善谱图质量. 近年来结合了非靶向和靶向方法优势的混合方法成为代谢组学领域新一代方法,本文分别综述了在单一和多个质谱平台上混合方法的开发,这种方法对实现代谢物高覆盖鉴定并保持良好定量性能的目标具有重大意义,但样品运行和数据筛选更为复杂,在多个仪器间的方法转换和参数优化的要求也更高,这种方法仍将不断被改进优化以更好地应用于代谢组学. 高覆盖代谢组学的发展为外暴露引起的生物毒性效应机制研究提供了方法手段,本文对其在环境毒理学领域的应用进行了综述,包括在重金属、有机污染物和联合毒性效应机制方面的应用进展.
LC-MS的技术提升无疑给高覆盖代谢物可靠检测带来了重大发展. 然而目前高覆盖代谢组学分析仍存在一些有待提高的方面,当前代谢组学提升的覆盖率仍然无法涵盖超痕量代谢物,大规模代谢物的准确分析仍然需要不断提升,且对于高通量样品分析需要更快速且准确性高的方法. 另外需要加强分析方法的标准化,从而有助于不同实验室和仪器间的比对,方法和数据的开放性也对进一步发展有重要推动作用. 在数据采集策略方面,这些问题的解决可以通过在质谱数据采集的速率、灵敏度、动态范围和准确性之间进行权衡,针对不同分析需求进行偏向性改进. 另外,机器学习的方法为采集策略的优化和后续的数据处理提供了新的思路.
-
图 1 分别在QQQ和Q-Orbitrap仪器上进行MRM(a)和PRM(b)的示意图
Figure 1. Schematic representation of MRM (a) and PRM (b) as performed on QQQ and Q-Orbitrap instrumentation respectively
-
[1] OLIVER S G, WINSON M K, KELL D B, et al. Systematic functional analysis of the yeast genome [J]. Trends in Biotechnology, 1998, 16(9): 373-378. doi: 10.1016/S0167-7799(98)01214-1 [2] NICHOLSON J K, WILSON I D. Understanding ‘global’ systems biology: Metabonomics and the continuum of metabolism [J]. Nature Reviews Drug Discovery, 2003, 2(8): 668-676. doi: 10.1038/nrd1157 [3] DUNN W B, BAILEY N J C, JOHNSON H E. Measuring the metabolome: Current analytical technologies [J]. The Analyst, 2005, 130(5): 606-625. doi: 10.1039/b418288j [4] FELL D A. Beyond genomics [J]. Trends in Genetics:TIG, 2001, 17(12): 680-682. doi: 10.1016/S0168-9525(01)02521-5 [5] FIEHN O. Metabolomics: the link between genotypes and phenotypes [J]. Plant Molecular Biology, 2002, 48(1/2): 155-171. doi: 10.1023/A:1013713905833 [6] WINDER C L, DUNN W B, SCHULER S, et al. Global metabolic profiling of Escherichia coli cultures: An evaluation of methods for quenching and extraction of intracellular metabolites [J]. Analytical Chemistry, 2008, 80(8): 2939-2948. doi: 10.1021/ac7023409 [7] BRUCE S J, TAVAZZI I, PARISOD V, et al. Investigation of human blood plasma sample preparation for performing metabolomics using ultrahigh performance liquid chromatography/mass spectrometry [J]. Analytical Chemistry, 2009, 81(9): 3285-3296. doi: 10.1021/ac8024569 [8] RICO E, GONZÁLEZ O, BLANCO M E, et al. Evaluation of human plasma sample preparation protocols for untargeted metabolic profiles analyzed by UHPLC-ESI-TOF-MS [J]. Analytical and Bioanalytical Chemistry, 2014, 406(29): 7641-7652. doi: 10.1007/s00216-014-8212-y [9] DETTMER K, NÜRNBERGER N, KASPAR H, et al. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: Optimization of harvesting and extraction protocols [J]. Analytical and Bioanalytical Chemistry, 2011, 399(3): 1127-1139. doi: 10.1007/s00216-010-4425-x [10] KIM S, LEE D Y, WOHLGEMUTH G, et al. Evaluation and optimization of metabolome sample preparation methods for Saccharomyces cerevisiae [J]. Analytical Chemistry, 2013, 85(4): 2169-2176. doi: 10.1021/ac302881e [11] JOHNSON C H, IVANISEVIC J, SIUZDAK G. Metabolomics: beyond biomarkers and towards mechanisms [J]. Nature Reviews Molecular Cell Biology, 2016, 17(7): 451-459. doi: 10.1038/nrm.2016.25 [12] WANT E J, NORDSTRÖM A, MORITA H, et al. From exogenous to endogenous: The inevitable imprint of mass spectrometry in metabolomics [J]. Journal of Proteome Research, 2007, 6(2): 459-468. doi: 10.1021/pr060505+ [13] MARX V. Boost that metabolomic confidence [J]. Nature Methods, 2020, 17(1): 33-36. doi: 10.1038/s41592-019-0694-2 [14] WISHART D S, FEUNANG Y D, MARCU A, et al. HMDB 4.0: The human metabolome database for 2018 [J]. Nucleic Acids Research, 2017, 46(D1): D608-D617. [15] ORTMAYR K, CAUSON T J, HANN S, et al. Increasing selectivity and coverage in LC-MS based metabolome analysis [J]. TrAC Trends in Analytical Chemistry, 2016, 82: 358-366. doi: 10.1016/j.trac.2016.06.011 [16] GIKA H G, WILSON I D, THEODORIDIS G A. LC-MS-based holistic metabolic profiling. Problems, limitations, advantages, and future perspectives [J]. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 2014, 966: 1-6. doi: 10.1016/j.jchromb.2014.01.054 [17] KITTERINGHAM N R, JENKINS R E, LANE C S, et al. Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics [J]. Journal of Chromatography B, 2009, 877(13): 1229-1239. doi: 10.1016/j.jchromb.2008.11.013 [18] CICCIMARO E, BLAIR I A. Stable-isotope dilution LC-MS for quantitative biomarker analysis [J]. Bioanalysis, 2010, 2(2): 311-341. doi: 10.4155/bio.09.185 [19] KOK M G M, NIX C, NYS G, et al. Targeted metabolomics of whole blood using volumetric absorptive microsampling [J]. Talanta, 2019, 197: 49-58. doi: 10.1016/j.talanta.2019.01.014 [20] BREIER M, WAHL S, PREHN C, et al. Targeted metabolomics identifies reliable and stable metabolites in human serum and plasma samples [J]. PLoS One, 2014, 9(2): e89728. doi: 10.1371/journal.pone.0089728 [21] KUHRING M, EISENBERGER A, SCHMIDT V, et al. Concepts and software package for efficient quality control in targeted metabolomics studies: MeTaQuaC [J]. Analytical Chemistry, 2020, 92(15): 10241-10245. doi: 10.1021/acs.analchem.0c00136 [22] MAZZINI F N, COOK F, GOUNARIDES J, et al. Plasma and stool metabolomic biomarkers of non-alcoholic fatty liver disease in Argentina [J]. MedRxiv, 2020, 8: 20165308. [23] DOMINGO-ALMENARA X, MONTENEGRO-BURKE J R, IVANISEVIC J, et al. XCMS-MRM and METLIN-MRM: A cloud library and public resource for targeted analysis of small molecules [J]. Nature Methods, 2018, 15(9): 681-684. doi: 10.1038/s41592-018-0110-3 [24] ZHENG F J, ZHAO X J, ZENG Z D, et al. Development of a plasma pseudotargeted metabolomics method based on ultra-high-performance liquid chromatography–mass spectrometry [J]. Nature Protocols, 2020, 15(8): 2519-2537. doi: 10.1038/s41596-020-0341-5 [25] ZHOU J T, LIU H Y, LIU Y, et al. Development and evaluation of a parallel reaction monitoring strategy for large-scale targeted metabolomics quantification [J]. Analytical Chemistry, 2016, 88(8): 4478-4486. doi: 10.1021/acs.analchem.6b00355 [26] SCHILLING B, MACLEAN B, HELD J M, et al. Multiplexed, scheduled, high-resolution parallel reaction monitoring on a full scan QqTOF instrument with integrated data-dependent and targeted mass spectrometric workflows [J]. Analytical Chemistry, 2015, 87(20): 10222-10229. doi: 10.1021/acs.analchem.5b02983 [27] TANG H, FANG H S, YIN E, et al. Multiplexed parallel reaction monitoring targeting histone modifications on the QExactive mass spectrometer [J]. Analytical Chemistry, 2014, 86(11): 5526-5534. doi: 10.1021/ac500972x [28] CHO K, SCHWAIGER-HABER M, NASER F J, et al. Targeting unique biological signals on the fly to improve MS/MS coverage and identification efficiency in metabolomics [J]. Analytica Chimica Acta, 2021, 1149: 338210. doi: 10.1016/j.aca.2021.338210 [29] CLANCY M V, ZYTYNSKA S E, MORITZ F, et al. Metabotype variation in a field population of tansy plants influences aphid host selection [J]. Plant, Cell & Environment, 2018, 41(12): 2791-2805. [30] MARR S, HAGEMAN J A, WEHRENS R, et al. LC-MS based plant metabolic profiles of thirteen grassland species grown in diverse neighbourhoods [J]. Scientific Data, 2021, 8: 52. doi: 10.1038/s41597-021-00836-8 [31] SMITH C A, WANT E J, O'MAILLE G, et al. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification [J]. Analytical Chemistry, 2006, 78(3): 779-787. doi: 10.1021/ac051437y [32] GUO J, HUAN T. Comparison of full-scan, data-dependent, and data-independent acquisition modes in liquid chromatography-mass spectrometry based untargeted metabolomics [J]. Analytical Chemistry, 2020, 92(12): 8072-8080. doi: 10.1021/acs.analchem.9b05135 [33] BENTON H P, IVANISEVIC J, MAHIEU N G, et al. Autonomous metabolomics for rapid metabolite identification in global profiling [J]. Analytical Chemistry, 2015, 87(2): 884-891. doi: 10.1021/ac5025649 [34] FENAILLE F, BARBIER SAINT-HILAIRE P, ROUSSEAU K, et al. Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? [J]. Journal of Chromatography A, 2017, 1526: 1-12. doi: 10.1016/j.chroma.2017.10.043 [35] DEFOSSEZ E, BOURQUIN J, von REUSS S, et al. Eight key rules for successful data-dependent acquisition in mass spectrometry-based metabolomics [J]. Mass Spectrometry Reviews, 2021, 7: 1-13. [36] BENDALL S C, HUGHES C, CAMPBELL J L, et al. An enhanced mass spectrometry approach reveals human embryonic stem cell growth factors in culture [J]. Molecular & Cellular Proteomics, 2009, 8(3): 421-432. [37] EDMANDS W M, FERRARI P, ROTHWELL J A, et al. Polyphenol metabolome in human urine and its association with intake of polyphenol-rich foods across European countries [J]. The American Journal of Clinical Nutrition, 2015, 102(4): 905-913. doi: 10.3945/ajcn.114.101881 [38] KOELMEL J P, KROEGER N M, GILL E L, et al. Expanding lipidome coverage using LC-MS/MS data-dependent acquisition with automated exclusion list generation [J]. Journal of the American Society for Mass Spectrometry, 2017, 28(5): 908-917. doi: 10.1007/s13361-017-1608-0 [39] ZHANG Z Q. Automated precursor ion exclusion during LC-MS/MS data acquisition for optimal ion identification [J]. Journal of the American Society for Mass Spectrometry, 2012, 23(8): 1400-1407. doi: 10.1007/s13361-012-0401-3 [40] HOOPMANN M R, MERRIHEW G E, von HALLER P D, et al. Post analysis data acquisition for the iterative MS/MS sampling of proteomics mixtures [J]. Journal of Proteome Research, 2009, 8(4): 1870-1875. doi: 10.1021/pr800828p [41] GINÉ R, CAPELLADES J, BADIA J M, et al. HERMES: a molecular-formula-oriented method to target the metabolome [J]. Nature Methods, 2021, 18(11): 1370-1376. doi: 10.1038/s41592-021-01307-z [42] AcquireX Intelligent Data Acquisition Workflow[C].[2022-02-08].https://www.thermofisher.cn/cn/zh/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/lc-ms-data-acquisition-software/acquirex-intelligent-data-acquisition-workflow.html.C].[2022--02-08].https://www.thermofisher.cn/cn/zh/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/lc-ms-data-acquisition-software/acquirex-intelligent-data-acquisition-workflow.html. [43] BROECKLING C D, HOYES E, RICHARDSON K, et al. Comprehensive tandem-mass-spectrometry coverage of complex samples enabled by data-set-dependent acquisition [J]. Analytical Chemistry, 2018, 90(13): 8020-8027. doi: 10.1021/acs.analchem.8b00929 [44] BILLS B, BARSHOP W D, SHARMA S, et al. Novel real-time library search driven data acquisition strategy for identification and characterization of metabolites [J]. Analytical Chemistry, 2022, 94(9): 3749-3755. doi: 10.1021/acs.analchem.1c04336 [45] WANDY J, DAVIES V, van der HOOFT J J, et al. In silico optimization of mass spectrometry fragmentation strategies in metabolomics [J]. Metabolites, 2019, 9(10): 219. doi: 10.3390/metabo9100219 [46] DAVIES V, WANDY J, WEIDT S, et al. Rapid development of improved data-dependent acquisition strategies [J]. Analytical Chemistry, 2021, 93(14): 5676-5683. doi: 10.1021/acs.analchem.0c03895 [47] CHEN L Y, ZHOU L, CHAN E C Y, et al. Characterization of the human tear metabolome by LC-MS/MS [J]. Journal of Proteome Research, 2011, 10(10): 4876-4882. doi: 10.1021/pr2004874 [48] SMITH C A, O'MAILLE G, WANT E J, et al. METLIN: a metabolite mass spectral database [J]. Therapeutic Drug Monitoring, 2005, 27(6): 747-751. doi: 10.1097/01.ftd.0000179845.53213.39 [49] HORAI H, ARITA M, KANAYA S, et al. MassBank: A public repository for sharing mass spectral data for life sciences [J]. Journal of Mass Spectrometry, 2010, 45(7): 703-714. doi: 10.1002/jms.1777 [50] SIEGEL D, MEINEMA A C, PERMENTIER H, et al. Integrated quantification and identification of aldehydes and ketones in biological samples [J]. Analytical Chemistry, 2014, 86(10): 5089-5100. doi: 10.1021/ac500810r [51] WRONA M, MAURIALA T, BATEMAN K P, et al. ‘All-in-One’ analysis for metabolite identification using liquid chromatography/hybrid quadrupole time-of-flight mass spectrometry with collision energy switching [J]. Rapid Communications in Mass Spectrometry, 2005, 19(18): 2597-2602. doi: 10.1002/rcm.2101 [52] PLUMB R S, JOHNSON K A, RAINVILLE P, et al. UPLC/MS(E);A new approach for generating molecular fragment information for biomarker structure elucidation [J]. Rapid Communications in Mass Spectrometry, 2006, 20(13): 1989-1994. doi: 10.1002/rcm.2550 [53] NAVARRO-REIG M, JAUMOT J, PIÑA B, et al. Metabolomic analysis of the effects of cadmium and copper treatment in Oryza sativa L. using untargeted liquid chromatography coupled to high resolution mass spectrometry and all-ion fragmentation [J]. Metallomics, 2017, 9(6): 660-675. doi: 10.1039/C6MT00279J [54] PERRY S J, NÁSZ S, SAEED M. A high-resolution accurate mass (HR/AM) approach to identification, profiling and characterization of in vitro nefazodone metabolites using a hybrid quadrupole Orbitrap (Q-Exactive) [J]. Rapid Communications in Mass Spectrometry, 2015, 29(17): 1545-1555. doi: 10.1002/rcm.7250 [55] SENTANDREU E, PERIS-DÍAZ M D, SWEENEY S R, et al. A survey of orbitrap all ion fragmentation analysis assessed by an R MetaboList package to study small-molecule metabolites [J]. Chromatographia, 2018, 81(7): 981-994. doi: 10.1007/s10337-018-3536-y [56] VENTURA G, BIANCO M, CALVANO C D, et al. HILIC-ESI-FTMS with all ion fragmentation (AIF) scans as a tool for fast lipidome investigations [J]. Molecules (Basel, Switzerland), 2020, 25(10): 2310. doi: 10.3390/molecules25102310 [57] van der LAAN T, BOOM I, MALIEPAARD J, et al. Data-independent acquisition for the quantification and identification of metabolites in plasma [J]. Metabolites, 2020, 10(12): 514. doi: 10.3390/metabo10120514 [58] PAGLIA G, ASTARITA G. Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry [J]. Nature Protocols, 2017, 12(4): 797-813. doi: 10.1038/nprot.2017.013 [59] BLAŽENOVIĆ I, KIND T, JI J, et al. Software tools and approaches for compound identification of LC-MS/MS data in metabolomics [J]. Metabolites, 2018, 8(2): 31. doi: 10.3390/metabo8020031 [60] GILLET L C, NAVARRO P, TATE S, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis [J]. Molecular & Cellular Proteomics, 2012, 11(6): O111.016717. [61] ZHANG Y, BILBAO A, BRUDERER T, et al. The use of variable Q1 isolation windows improves selectivity in LC-SWATH-MS acquisition [J]. Journal of Proteome Research, 2015, 14(10): 4359-4371. doi: 10.1021/acs.jproteome.5b00543 [62] AKBAL L, HOPFGARTNER G. Supercritical fluid chromatography-mass spectrometry using data independent acquisition for the analysis of polar metabolites in human urine [J]. Journal of Chromatography A, 2020, 1609: 460449. doi: 10.1016/j.chroma.2019.460449 [63] BONNER R, HOPFGARTNER G. SWATH acquisition mode for drug metabolism and metabolomics investigations [J]. Bioanalysis, 2016, 8(16): 1735-1750. doi: 10.4155/bio-2016-0141 [64] BONNER R, HOPFGARTNER G. SWATH data independent acquisition mass spectrometry for metabolomics [J]. TrAC Trends in Analytical Chemistry, 2019, 120: 115278. doi: 10.1016/j.trac.2018.10.014 [65] WANG R H, YIN Y D, ZHU Z J. Advancing untargeted metabolomics using data-independent acquisition mass spectrometry technology [J]. Analytical and Bioanalytical Chemistry, 2019, 411(19): 4349-4357. doi: 10.1007/s00216-019-01709-1 [66] XIONG Y T, SHI C, ZHONG F, et al. LC-MS/MS and SWATH based serum metabolomics enables biomarker discovery in pancreatic cancer [J]. Clinica Chimica Acta, 2020, 506: 214-221. doi: 10.1016/j.cca.2020.03.043 [67] XU L, XU Z Z, STRASHNOV I, et al. Use of information dependent acquisition mass spectra and sequential window acquisition of all theoretical fragment-ion mass spectra for fruit juices metabolomics and authentication [J]. Metabolomics, 2020, 16(7): 81. doi: 10.1007/s11306-020-01701-2 [68] MOSELEY M A, HUGHES C J, JUVVADI P R, et al. Scanning quadrupole data-independent acquisition, part A: Qualitative and quantitative characterization [J]. Journal of Proteome Research, 2018, 17(2): 770-779. doi: 10.1021/acs.jproteome.7b00464 [69] MESSNER C, DEMICHEV V, BLOOMFIELD N, et al. ScanningSWATH enables ultra-fast proteomics using high-flow chromatography and minute-scale gradients[J]. BioRxiv, 2019,DOI:10.1101/656793. [70] MUN D G, RENUSE S, SARASWAT M, et al. PASS-DIA: A data-independent acquisition approach for discovery studies [J]. Analytical Chemistry, 2020, 92(21): 14466-14475. doi: 10.1021/acs.analchem.0c02513 [71] GUO J, SHEN S, XING S P, et al. DaDIA: hybridizing data-dependent and data-independent acquisition modes for generating high-quality metabolomic data [J]. Analytical Chemistry, 2021, 93(4): 2669-2677. doi: 10.1021/acs.analchem.0c05022 [72] CHAPMAN J D, GOODLETT D R, MASSELON C D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling [J]. Mass Spectrometry Reviews, 2014, 33(6): 452-470. doi: 10.1002/mas.21400 [73] TADA I, CHALECKIS R, TSUGAWA H, et al. Correlation-based deconvolution (CorrDec) to generate high-quality MS2 spectra from data-independent acquisition in multisample studies [J]. Analytical Chemistry, 2020, 92(16): 11310-11317. doi: 10.1021/acs.analchem.0c01980 [74] TSUGAWA H, CAJKA T, KIND T, et al. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis [J]. Nature Methods, 2015, 12(6): 523-526. doi: 10.1038/nmeth.3393 [75] YIN Y D, WANG R H, CAI Y P, et al. DecoMetDIA: deconvolution of multiplexed MS/MS spectra for metabolite identification in SWATH-MS-based untargeted metabolomics [J]. Analytical Chemistry, 2019, 91(18): 11897-11904. doi: 10.1021/acs.analchem.9b02655 [76] LI H, CAI Y P, GUO Y, et al. MetDIA: targeted metabolite extraction of multiplexed MS/MS spectra generated by data-independent acquisition [J]. Analytical Chemistry, 2016, 88(17): 8757-8764. doi: 10.1021/acs.analchem.6b02122 [77] XUE J C, GUIJAS C, BENTON H P, et al. METLIN MS2 molecular standards database: A broad chemical and biological resource [J]. Nature Methods, 2020, 17(10): 953-954. doi: 10.1038/s41592-020-0942-5 [78] WANG M X, CARVER J J, PHELAN V V, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking [J]. Nature Biotechnology, 2016, 34(8): 828-837. doi: 10.1038/nbt.3597 [79] CHEN S L, KONG H W, LU X, et al. Pseudotargeted metabolomics method and its application in serum biomarker discovery for hepatocellular carcinoma based on ultra high-performance liquid chromatography/triple quadrupole mass spectrometry [J]. Analytical Chemistry, 2013, 85(17): 8326-8333. doi: 10.1021/ac4016787 [80] CHEN Y H, XU J, ZHANG R P, et al. Assessment of data pre-processing methods for LC-MS/MS-based metabolomics of uterine cervix cancer [J]. The Analyst, 2013, 138(9): 2669-2677. doi: 10.1039/c3an36818a [81] CHEN L, ZHONG F Y, ZHU J J. Bridging targeted and untargeted mass spectrometry-based metabolomics via hybrid approaches [J]. Metabolites, 2020, 10(9): 348. doi: 10.3390/metabo10090348 [82] WANG Y, LIU F, LI P, et al. An improved pseudotargeted metabolomics approach using multiple ion monitoring with time-staggered ion lists based on ultra-high performance liquid chromatography/quadrupole time-of-flight mass spectrometry [J]. Analytica Chimica Acta, 2016, 927: 82-88. doi: 10.1016/j.aca.2016.05.008 [83] GU H W, ZHANG P, ZHU J J, et al. Globally optimized targeted mass spectrometry: Reliable metabolomics analysis with broad coverage [J]. Analytical Chemistry, 2015, 87(24): 12355-12362. doi: 10.1021/acs.analchem.5b03812 [84] SHI X J, WANG S, JASBI P, et al. Database-assisted globally optimized targeted mass spectrometry (dGOT-MS): Broad and reliable metabolomics analysis with enhanced identification [J]. Analytical Chemistry, 2019, 91(21): 13737-13745. doi: 10.1021/acs.analchem.9b03107 [85] GUIJAS C, MONTENEGRO-BURKE J R, DOMINGO-ALMENARA X, et al. METLIN: A technology platform for identifying knowns and unknowns [J]. Analytical Chemistry, 2018, 90(5): 3156-3164. doi: 10.1021/acs.analchem.7b04424 [86] ZHONG F Y, XU M Y, ZHU J J. Development and application of time staggered/mass staggered-globally optimized targeted mass spectrometry [J]. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 2019, 1120: 80-88. doi: 10.1016/j.jchromb.2019.04.051 [87] GAO Y, CHEN Y H, YUE X F, et al. Development of simultaneous targeted metabolite quantification and untargeted metabolomics strategy using dual-column liquid chromatography coupled with tandem mass spectrometry [J]. Analytica Chimica Acta, 2018, 1037: 369-379. doi: 10.1016/j.aca.2018.08.042 [88] ZHANG L, ZHENG W, LI X, et al. A merged method for targeted analysis of amino acids and derivatives using parallel reaction monitoring combined with untargeted profiling by HILIC-Q-Orbitrap HRMS [J]. Journal of Pharmaceutical and Biomedical Analysis, 2021, 203: 114208. doi: 10.1016/j.jpba.2021.114208 [89] LI Y, RUAN Q, LI Y L, et al. A novel approach to transforming a non-targeted metabolic profiling method to a pseudo-targeted method using the retention time locking gas chromatography/mass spectrometry-selected ions monitoring [J]. Journal of Chromatography A, 2012, 1255: 228-236. doi: 10.1016/j.chroma.2012.01.076 [90] LUO P, DAI W D, YIN P Y, et al. Multiple reaction monitoring-ion pair finder: A systematic approach to transform nontargeted mode to pseudotargeted mode for metabolomics study based on liquid chromatography-mass spectrometry [J]. Analytical Chemistry, 2015, 87(10): 5050-5055. doi: 10.1021/acs.analchem.5b00615 [91] ZHA H H, CAI Y P, YIN Y D, et al. SWATHtoMRM: development of high-coverage targeted metabolomics method using SWATH technology for biomarker discovery [J]. Analytical Chemistry, 2018, 90(6): 4062-4070. doi: 10.1021/acs.analchem.7b05318 [92] CHEN Y H, ZHOU Z, YANG W, et al. Development of a data-independent targeted metabolomics method for relative quantification using liquid chromatography coupled with tandem mass spectrometry [J]. Analytical Chemistry, 2017, 89(13): 6954-6962. doi: 10.1021/acs.analchem.6b04727 [93] LUO P, YIN P Y, HUA R, et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma [J]. Hepatology (Baltimore, Md. ), 2018, 67(2): 662-675. doi: 10.1002/hep.29561 [94] SHAO Y P, ZHU B, ZHENG R Y, et al. Development of urinary pseudotargeted LC-MS-based metabolomics method and its application in hepatocellular carcinoma biomarker discovery [J]. Journal of Proteome Research, 2015, 14(2): 906-916. doi: 10.1021/pr500973d [95] WANG L C, SU B Z, ZENG Z D, et al. Ion-pair selection method for pseudotargeted metabolomics based on SWATH MS acquisition and its application in differential metabolite discovery of type 2 diabetes [J]. Analytical Chemistry, 2018, 90(19): 11401-11408. doi: 10.1021/acs.analchem.8b02377 [96] ZHOU Y, SONG R X, MA C, et al. Discovery and validation of potential urinary biomarkers for bladder cancer diagnosis using a pseudotargeted GC-MS metabolomics method [J]. Oncotarget, 2017, 8(13): 20719-20728. doi: 10.18632/oncotarget.14988 [97] FANG C N, SU B Z, JIANG T Y, et al. Prognosis prediction of hepatocellular carcinoma after surgical resection based on serum metabolic profiling from gas chromatography-mass spectrometry [J]. Analytical and Bioanalytical Chemistry, 2021, 413(12): 3153-3165. doi: 10.1007/s00216-021-03281-z [98] SUN H Q, CHEN N, WANG X C, et al. The study on the pathogenesis of pediatric lymphoma based on the combination of pseudotargeted and targeted metabolomics [J]. BioMed Research International, 2021, 2021: 9984357. [99] YE G Z, LIU Y, YIN P Y, et al. Study of induction chemotherapy efficacy in oral squamous cell carcinoma using pseudotargeted metabolomics [J]. Journal of Proteome Research, 2014, 13(4): 1994-2004. doi: 10.1021/pr4011298 [100] ZHAO Y N, ZHAO C X, LU X, et al. Investigation of the relationship between the metabolic profile of tobacco leaves in different planting regions and climate factors using a pseudotargeted method based on gas chromatography/mass spectrometry [J]. Journal of Proteome Research, 2013, 12(11): 5072-5083. doi: 10.1021/pr400799a [101] ZHAO Y N, ZHANG L, ZHAO C X, et al. Metabolic responses of rice leaves and seeds under transgenic backcross breeding and pesticide stress by pseudotargeted metabolomics [J]. Metabolomics, 2015, 11(6): 1802-1814. doi: 10.1007/s11306-015-0834-3 [102] ZHANG J, WU X F, QIU J Q, et al. Comprehensive comparison on the chemical profile of Guang Chen pi at different ripeness stages using untargeted and pseudotargeted metabolomics [J]. Journal of Agricultural and Food Chemistry, 2020, 68(31): 8483-8495. doi: 10.1021/acs.jafc.0c02904 [103] LI L L, WANG Y, LI Y X, et al. A pseudotargeted method based on sequential window acquisition of all theoretical spectra mass spectrometry acquisition and its application in quality assessment of traditional Chinese medicine preparation-Yuanhu Zhitong Tablet [J]. Journal of Separation Science, 2022, 45(2): 650-658. doi: 10.1002/jssc.202100611 [104] XIAO J M, SONG J Y, SA Y H, et al. The mechanisms of improving IVF outcomes of Liu-Wei-di-Huang pill acting on DOR patients [J]. Evidence-Based Complementary and Alternative Medicine, 2020, 2020: 5183017. [105] WANG F D, ZHANG H J, GENG N B, et al. A metabolomics strategy to assess the combined toxicity of polycyclic aromatic hydrocarbons (PAHs) and short-chain chlorinated paraffins (SCCPs) [J]. Environmental Pollution, 2018, 234: 572-580. doi: 10.1016/j.envpol.2017.11.073 [106] WANG F D, ZHANG H J, GENG N B, et al. New insights into the cytotoxic mechanism of hexabromocyclododecane from a metabolomic approach [J]. Environmental Science & Technology, 2016, 50(6): 3145-3153. [107] LIU D Y, YANG J N, JIN W B, et al. A high coverage pseudotargeted lipidomics method based on three-phase liquid extraction and segment data-dependent acquisition using UHPLC-MS/MS with application to a study of depression rats [J]. Analytical and Bioanalytical Chemistry, 2021, 413(15): 3975-3986. doi: 10.1007/s00216-021-03349-w [108] XU S L, LV X, WU B F, et al. Pseudotargeted lipidomics strategy enabling comprehensive profiling and precise lipid structural elucidation of polyunsaturated lipid-rich Echium oil [J]. Journal of Agricultural and Food Chemistry, 2021, 69(32): 9012-9024. doi: 10.1021/acs.jafc.0c07268 [109] XUAN Q H, HU C X, YU D, et al. Development of a high coverage pseudotargeted lipidomics method based on ultra-high performance liquid chromatography-mass spectrometry [J]. Analytical Chemistry, 2018, 90(12): 7608-7616. doi: 10.1021/acs.analchem.8b01331 [110] LUO P, YIN P Y, ZHANG W J, et al. Optimization of large-scale pseudotargeted metabolomics method based on liquid chromatography-mass spectrometry [J]. Journal of Chromatography A, 2016, 1437: 127-136. doi: 10.1016/j.chroma.2016.01.078 [111] ZHAO J J, GUO X M, WANG X C, et al. A chemometric strategy to automatically screen selected ion monitoring ions for gas chromatography-mass spectrometry-based pseudotargeted metabolomics [J]. Journal of Chromatography A, 2022, 1664: 462801. doi: 10.1016/j.chroma.2021.462801 [112] MOUNICOU S, SZPUNAR J, LOBINSKI R. Metallomics: the concept and methodology [J]. Chemical Society Reviews, 2009, 38(4): 1119-1138. doi: 10.1039/b713633c [113] CHEN C J, HSUEH Y M, LAI M S, et al. Increased prevalence of hypertension and long-term arsenic exposure [J]. Hypertension, 1995, 25(1): 53-60. doi: 10.1161/01.HYP.25.1.53 [114] FARZAN S F, CHEN Y, WU F, et al. Blood pressure changes in relation to arsenic exposure in a US pregnancy cohort [J]. Environmental Health Perspectives, 2015, 123(10): 999-1006. doi: 10.1289/ehp.1408472 [115] NAVAS-ACIEN A. Arsenic exposure and prevalence of type 2 diabetes in US adults [J]. JAMA, 2008, 300(7): 814. doi: 10.1001/jama.300.7.814 [116] WANG X X, MU X L, ZHANG J, et al. Serum metabolomics reveals that arsenic exposure disrupted lipid and amino acid metabolism in rats: A step forward in understanding chronic arsenic toxicity [J]. Metallomics, 2015, 7(3): 544-552. doi: 10.1039/C5MT00002E [117] LI H, WANG M, LIANG Q D, et al. Urinary metabolomics revealed arsenic exposure related to metabolic alterations in general Chinese pregnant women [J]. Journal of Chromatography A, 2017, 1479: 145-152. doi: 10.1016/j.chroma.2016.12.007 [118] SPRATLEN M J, GRAU-PEREZ M, UMANS J G, et al. Targeted metabolomics to understand the association between arsenic metabolism and diabetes-related outcomes: Preliminary evidence from the Strong Heart Family Study [J]. Environmental Research, 2019, 168: 146-157. doi: 10.1016/j.envres.2018.09.034 [119] CHEN S, ZHANG M Y, BO L, et al. Metabolomic analysis of the toxic effect of chronic exposure of cadmium on rat urine [J]. Environmental Science and Pollution Research International, 2018, 25(4): 3765-3774. doi: 10.1007/s11356-017-0774-8 [120] ZENG T, LIANG Y S, CHEN J Y, et al. Urinary metabolic characterization with nephrotoxicity for residents under cadmium exposure [J]. Environment International, 2021, 154: 106646. doi: 10.1016/j.envint.2021.106646 [121] EDWARDS M. Fetal death and reduced birth rates associated with exposure to lead-contaminated drinking water [J]. Environmental Science & Technology, 2014, 48(1): 739-746. [122] MIELKE H W, ZAHRAN S. The urban rise and fall of air lead (Pb) and the latent surge and retreat of societal violence [J]. Environment International, 2012, 43: 48-55. doi: 10.1016/j.envint.2012.03.005 [123] EGUCHI A, NOMIYAMA K, SAKURAI K, et al. Alterations in urinary metabolomic profiles due to lead exposure from a lead-acid battery recycling site [J]. Environmental Pollution, 2018, 242: 98-105. doi: 10.1016/j.envpol.2018.06.071 [124] KELLY R S, BAYNE H, SPIRO A II, et al. Metabolomic signatures of lead exposure in the VA Normative Aging Study [J]. Environmental Research, 2020, 190: 110022. doi: 10.1016/j.envres.2020.110022 [125] SUN R L, XU K, JI S B, et al. Benzene exposure induces gut microbiota dysbiosis and metabolic disorder in mice [J]. Science of the Total Environment, 2020, 705: 135879. doi: 10.1016/j.scitotenv.2019.135879 [126] JI H N, SONG N N, REN J, et al. Metabonomics reveals bisphenol A affects fatty acid and glucose metabolism through activation of LXR in the liver of male mice [J]. Science of the Total Environment, 2020, 703: 134681. doi: 10.1016/j.scitotenv.2019.134681 [127] LAW K L, THOMPSON R C. Microplastics in the seas [J]. Science, 2014, 345(6193): 144-145. doi: 10.1126/science.1254065 [128] HUANG W, WANG X H, CHEN D Y, et al. Toxicity mechanisms of polystyrene microplastics in marine mussels revealed by high-coverage quantitative metabolomics using chemical isotope labeling liquid chromatography mass spectrometry [J]. Journal of Hazardous Materials, 2021, 417: 126003. doi: 10.1016/j.jhazmat.2021.126003 [129] YANG L X, LIU Y P, CUI Z, et al. Metabolomic mechanisms of short chain chlorinated paraffins toxicity in rats [J]. Environmental Research, 2021, 197: 111060. doi: 10.1016/j.envres.2021.111060 [130] WANG Z H, ZHENG Y J, ZHAO B X, et al. Human metabolic responses to chronic environmental polycyclic aromatic hydrocarbon exposure by a metabolomic approach [J]. Journal of Proteome Research, 2015, 14(6): 2583-2593. doi: 10.1021/acs.jproteome.5b00134 [131] MIDGETT K, PEDEN-ADAMS M M, GILKESON G S, et al. In vitro evaluation of the effects of perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) on IL-2 production in human T-cells [J]. Journal of Applied Toxicology, 2015, 35(5): 459-465. doi: 10.1002/jat.3037 [132] LI R, GUO C, TSE W K F, et al. Metabolomic analysis reveals metabolic alterations of human peripheral blood lymphocytes by perfluorooctanoic acid [J]. Chemosphere, 2020, 239: 124810. doi: 10.1016/j.chemosphere.2019.124810 [133] XU M Y, WANG P, SUN Y J, et al. Metabolomic analysis for combined hepatotoxicity of chlorpyrifos and cadmium in rats [J]. Toxicology, 2017, 384: 50-58. doi: 10.1016/j.tox.2017.04.008 [134] LIU F F, CHEN X L, LIU Y S, et al. Serum cardiovascular-related metabolites disturbance exposed to different heavy metal exposure scenarios [J]. Journal of Hazardous Materials, 2021, 415: 125590. doi: 10.1016/j.jhazmat.2021.125590 期刊类型引用(2)
1. 黄鹏,张静,王鸿辉,井江心,曾燕琼,陈铮,刘洋之. 腐植酸负载纳米零价铁对镉污染农田水稻籽粒代谢的影响. 农业环境科学学报. 2024(04): 732-740 .  百度学术
百度学术
2. 原晓喻,葛贝宁,徐丽娜,李熠晨,田海宁,余小领,睢福庆,刘炳杉. 环境中橡胶防老剂6PPD及其转化物检测方法. 环境化学. 2024(12): 3976-3989 . 本站查看
其他类型引用(0)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 6989
- HTML全文浏览数: 6989
- PDF下载数: 147
- 施引文献: 2
