













-
抗生素是一类具有抵抗微生物活性的天然、半合成或人工合成的化合物[1-2],能够干扰或抑制致病微生物的存在,在人和动物的疾病治疗,作为畜禽的饲料添加剂,促进动物生长等方面具有重要作用. 我国是抗生素的生产和使用大国,年均使用量约2.5万t,占全球总使用量的20%左右[3-4],但由于大量工业废水和生活污水未得到及时有效的处理便排放到环境中,且抗生素不易被人或动物吸收利用,易诱导“超级细菌”的出现,因此具有较高的生态和人类健康风险[5]. 磺胺嘧啶(sulfadiazine,SD)作为最常用的磺胺类抗生素药物之一,具有稳定性高、亲水性强的特点,在沉积物、地表水和地下水广泛存在[6,1-8]. 部分研究已证实磺胺类药物对人体健康有较大危害且广泛存在于环境中,因此对抗生素的治理势在必行.
在各类抗生素的处理技术中,高级氧化技术是最具有应用前景的技术. 电化学氧化技术因操作简单,工艺条件温和,无二次污染,反应活性高,逐渐成为抗生素废水处理中可行性最高的技术. 在电化学技术中,掺硼金刚石膜(boron-doped diamond,BDD)电极因具有电化学窗口宽、背景电流低、抗腐蚀性强、化学性能稳定等诸多优点在电化学氧化系统中得到广泛应用[9]. 在以BDD为阳极,碳毡(carbon felt,CF)为阴极的电化学氧化系统中,阳极BDD产生·OH,亲电进攻阳极附近的有机污染物. 阴极CF可在原位生成H2O2,在Fe2+存在的条件下,H2O2 被催化分解产生大量·OH,构成经典的电芬顿反应. 但在室温和室压下,氧气在水中的低溶解度(约8 mg·L−1)直接影响了空气中的氧气在传统阴极上的氧传质[10],导致空气氧源的利用率非常低[11]. 因此,气体扩散电极 (Gas diffusion electrode,GDE)被广泛研究以提高阴极催化层氧传质效率,它允许氧气从外部供应到阴极表面,而无需将其溶解在电解质中并提高 H2O2 电合成效率. 雷阳明等[12]制备出的GDE的效果远优于石墨电极,H2O2生成量为石墨电极的10倍,酸性红B溶液在阴极室的脱色率和COD去除率分别为94.2%和66.8%. 周明华等[13]以碳毡为基底制备的自然空气扩散电极 (natural air diffusion electrode,NADE),与普通气体扩散电极(GDE)系统相比,阴极的氧扩散系数提高了约 5.7倍,使 H2O2 产量显着增加(101.67 mg·h−1·cm−2). 孙秀萍等[11]研究了GDE的不同氧源的贡献率,发现H2O2的积累规律为双氧源模式>空气氧源>阳极析氧.
近年来,以活化过硫酸盐(persulfate, PS)产生硫酸根自由基(·SO4−)作为主要强氧化性基团降解有机污染物的新型高级氧化技术发展迅速,Fe2+作为催化剂活化PS进而氧化污染物逐渐成为研究热点,研究发现Fe2+活化 PS 能有效降解CIP、SMX、左氧氟沙星和TMP等抗生素[14-15]. 而研究表明[16],在BDD系统中,作为电解质的Na2SO4会在阳极反应生成一定量的S2O82−,这为电芬顿和过硫酸盐活化相结合提供新思路.
本文通过在阳极阴极之间加入质子交换膜的方式,分别构建单室反应器和阴阳极分隔的双室反应器,以SD作为目标污染物,探索系统对磺胺类抗生素的去除效率并明确该系统的作用机制.
-
BDD电极,德国CONDIAS GmbH公司;碳毡电极,天津碳素厂;15 mm×20 mm×1.5 mm不锈钢电极,购自上海越磁电子科技有限公司;Nafion115膜,10 cm×10 cm,购自杜邦Dupont.
-
还原性铁粉,分析纯,购自天津市鼎盛鑫化工有限公司;甲醇(CH3OH),色谱纯(>99.9%),购自北京迈瑞达科技有限公司;浓硫酸(H2SO4)(>98%),无水硫酸钠(Na2SO4)、氢氧化钠(NaOH)均为分析纯,购自国药集团化学试剂有限公司;草酸钛钾二水合物(C4K2O9Ti·2H2O),分析纯,购自罗恩试剂;聚四氟乙烯(PTFE),质量分数为60%,购自麦克林;磺胺嘧啶(C10H10N4O2S)(>98%),购自山东西亚化学股份有限公司.
-
FA1004电子天平;DZF-6020真空干燥箱;JB-1A磁力搅拌器;DH1765-1型程控直流稳压稳流电源;KQ-100DE数控超声波清洗器;日本岛津LC-20A高效液相色谱仪;安捷伦SB-C18(4.6 mm×250 mm×5 μm)色谱柱;紫外可见分光光度计(V-750),日本JASCO公司;QE Plus超高效液相色谱串联高分辨质谱仪;马弗炉,上海西马格高温电炉有限公司.
-
根据周明华[13]等的研究制备气体扩散阴极NADE,将CF依次浸于超纯水、乙醇、超纯水中超声清洗10 min,泡入2% PTFE中10 min,晾干,于马弗炉中360 ℃煅烧30 min. 定量取质量比为1:1的炭黑和PTFE(60%),于少量去离子水中超声并搅拌45 min,均匀涂覆于处理后的CF一侧,晾干. 用毛笔蘸取PTFE(60%)于涂有炭黑的一面继续涂覆5层,放入马弗炉中360 ℃煅烧30 min.
-
电化学氧化实验中设置3种系统对比降解效果. 对照组阳极为BDD电极,阴极分别为不锈电极(Stainless steel electrodes,SS)和NADE电极,两电极板平行放置,间距约1.5 cm,电极板面积均为1.25 cm2. 电解液为30 mg·L−1 SD + 0.1 mol·L−1 Na2SO4,电解液体积为250 mL,外加电流60 mA,温度为(23±1)℃,电解时间6 h,pH=3,pH用H2SO4和NaOH调节. 实验组阳极为BDD电极,阴极为CF电极,还原性Fe粉投加量为0.02 g,其余条件与对照组一致. 电催化氧化实验过程中通过磁力搅拌使溶液充分混合均匀,转速为15 r·s−1.
-
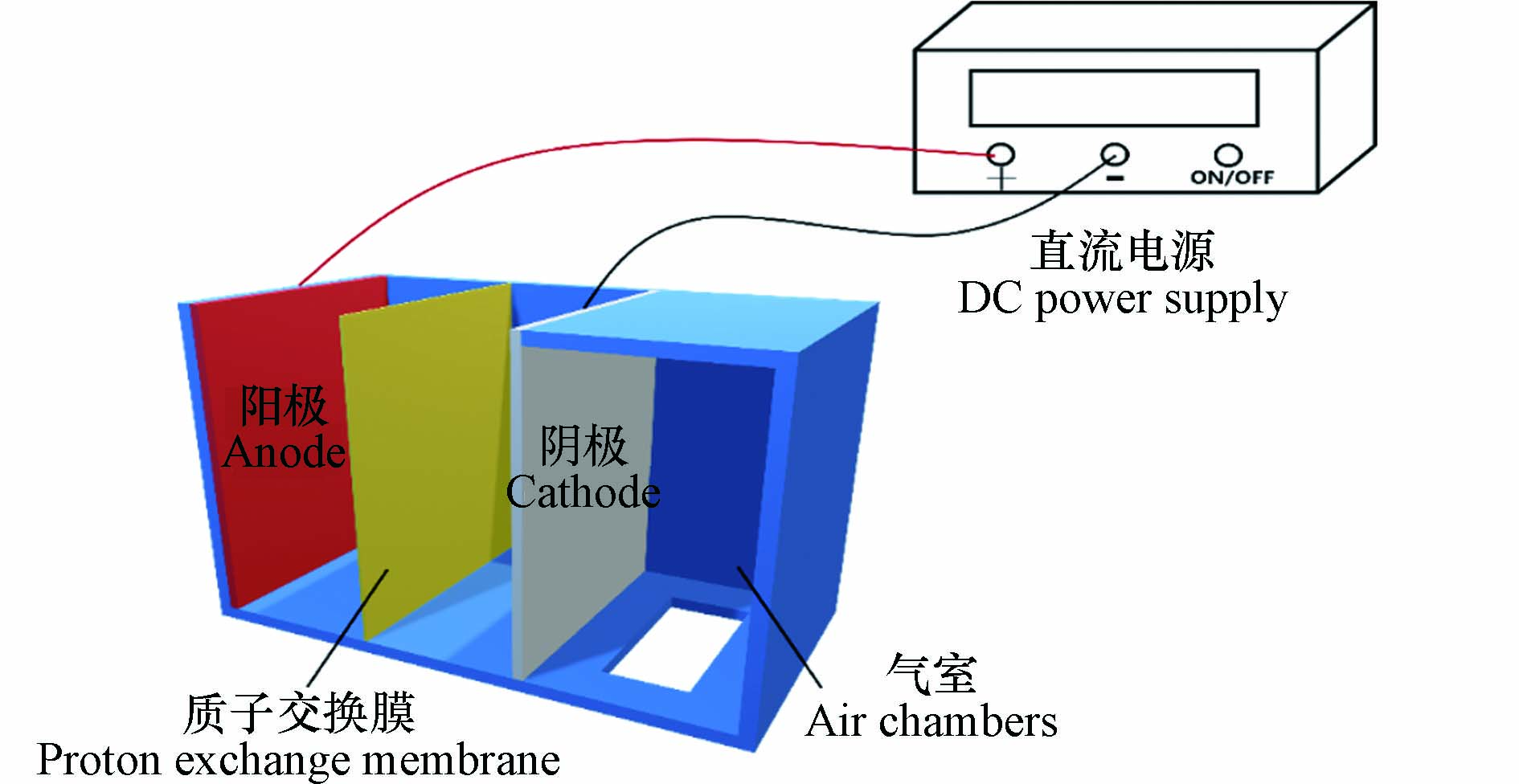
如图1所示,电解装置为定制的阴阳极分槽反应器,阳、阴极室有效容积均为324 mL,以阳离子交换膜(Nafion115)分隔;BDD阳极板和相同面积CF阴极板竖直平行放置,浸没高度为40 mm,间距为15 mm,电解液由曝气泵通入空气进行搅拌,电极连接直流稳压稳流电源,通过铜制导线与电极夹连接,实验中以30 mA·cm−2电流密度恒电流供电.根据对照情况配置不同电解质溶液 125 mL 分别装入阴阳极室,设置取样间隔时间为 30 min.
-
采用钛盐光度法测定反应过程中H2O2的浓度. 酸性条件下TiO2+能与H2O2形成稳定的黄色络合物[TiO(H2O2)]2+. 配制浓度为0.01 mol·L−1 H2O2,精确移取0、0.1、0.2、0.3、0.4、0.5 mL分别放入10 mL具塞比色管中,投加1 mL浓度为0.05 mol·L−1草酸钛钾作为显色剂和2 mL 3 mol·L−1硫酸溶液使体系保持一定酸度以消除杂离子干扰. 显色10 min后,在最大吸收波长400 nm下测定吸光度值.
采用基于改良碘量法的紫外-可见分光光度法测定过硫酸盐. 4 g KI加入到装有40 mL超纯水的锥形瓶中,摇匀. 配制浓度为0.02 mol·L−1 PS,精确移取0、0.2、0.4、0.6、0.8、1 mL分别放入10 mL具塞比色管中,投加1 mL KI作为显色剂,显色15 min后,在最大吸收波长352 nm下测定其吸光度值.
实验选用甲醇(Methanol,MeOH)对·OH和·SO4−进行淬灭,在含有污染物的电解液中加入过量的MeOH,尽可能淬灭阴、阳极产生·OH和·SO4−,其他条件不变,测定污染物的降解率. 此外,实验中将电解质0.1 mol·L−1 Na2SO4替换为0.2 mol·L−1 NaNO3,在保证电导率在较小范围内波动的同时,该系统内不会产生S2O82−和·SO4−. 其他条件不变,测定污染物的降解率,分析·OH和·SO4−对降解的贡献.
利用Gaussian 09软件和密度泛函理论(density functional theory,DFT)在B3LYP/6-31+g(d,p)水平下优化SD的几何结构,并计算SD分子中各原子的福井函数与电子云密度,结合两个值的大小判断SD分子中的反应活性位点.
使用高效液相色谱串联质谱(liquid chromatograph mass spectrometer,LC-MS)测定SD降解中间产物,实验中使用QE Plus超高效液相色谱串联高分辨质谱仪对SD降解过程中0.5、1、2、3、4、5、6 h时间点所取样品进行中间产物测定,采用Waters X Bridge C18(3.0 mm×150 mm,3.5 μm),样品过0.22 μm滤膜后进样分析. 在正离子模式下质谱条件:进样量5 μL;离子源类型:电喷雾离子源(Electron Spray Ionization,ESI);喷雾电压3500 V,雾化温度500 ℃;扫描范围50—1200. 所得质谱图用于分析SD降解过程中的中间产物,并结合上一步骤中的计算结果分析SD降解机制.
-
如图2,在单一电解室中,以BDD为阳极,分别以不锈钢电极(stainless steel, SS)、NADE为阴极,对比以下4种不同体系中的SD降解效果. 从图2可以看出,BDD-Fe-NADE系统展现了显著的降解优势. BDD-SS系统中,起主要降解作用的是阳极产生的·OH,180 min时降解率达52%. BDD-NADE系统中,除阳极产生·OH外,O2在阴极被还原成H2O2在阳极被激活产生·OH也对SD有降解作用,因此相对BDD-SS系统效果有提升,180 min时降解率达73%. BDD-Fe-SS系统中,相比BDD-SS系统降解效果更好,180 min时降解率达87%可能是因为除·OH外,SO42−在阳极被氧化为S2O82−,而后被Fe2+激活产生·SO4−,以降解SD. BDD-Fe-NADE系统的效果最好,60 min时SD几乎降解完全,可能是因为零价Fe在酸性溶液中可以析出Fe2+,从而激活阳极产生的S2O82−和阴极产生的H2O2,产生更多氧化性粒子,大大提升了降解效果.
-
在阴、阳极室中各加入100 mL浓度为30 mg·L−1的SD溶液,控制反应过程中的pH保持在3左右,在以下不同的实验条件下进行降解,利用高效液相色谱法(HPLC)测定SD的降解效果,并用一级动力学模型对其结果进行拟合,结果如图3所示.
从图3a、b中可以看出,在阳极室中,用MeOH淬灭后的降解效果最差,但仍有一定的降解效果,说明BDD作为阳极可以通过直接的电子传递(direct electron transfer,DET)氧化SD. 在NaNO3做电解质的系统中,阳极室中的SD除了通过DET降解外,还可被BDD表面通过产生的·OH氧化,180 min时降解率达56%. 在Na2SO4做电解质且不外加Fe的系统中,重要氧化途径仍为的为DET和·OH氧化,但由于SO42−会在BDD表面通过电子转移和间接氧化两个途径转化为S2O82-[17],少部分S2O82−在电激活的状态下也具有一定的氧化性能[18],因此该系统的降解效果较NaNO3做电解质的系统好. 在加入0.01g Fe后,系统的降解效果有了较为明显的提升,180 min时的降解率达到了86%,是因为加入的Fe在系统中析出的Fe2+可以激活积累的S2O82−转化为具有较强氧化性的·SO4−,提高了阳极室的降解率.
从图3c、d中可以看出,在阴极室中,用MeOH淬灭后和不加MeOH淬灭的降解效果相近,且均无明显的降解,说明在阴极上SD基本上不通过电子传递被氧化,且阴极室积累的H2O2也没有体现好的降解性,其可能原因是低浓度的H2O2氧化性较差. 在加入0.01g Fe后,其降解效果有了显著的提升,120 min时已将阴极室中的SD几乎降解完全,是因为Fe在酸性条件下析出的Fe2+可以激活积累的H2O2转化为具有较强氧化性的·OH,大大地提升了阴极室的氧化能力.
-
以上实验条件下的反应过程可符合一级动力学反应,如表1为阴、阳极室中不同条件降解效果的一级动力学模型,可通过建立不同实验条件下的一级动力学模型来分析不同氧化性物种在降解SD过程中的贡献,其计算结果如表2所示.
从表1的拟合曲线可以得到不同实验条件下的反应速率,通过分析不同系统中起主要作用的氧化性物种,以反应速率差值来表征氧化性物种的相对贡献率,可计算得到表2的结果. 从图4可以清晰地看到,在BDD-Fe-NADE系统中,阴极室起主要氧化作用的粒子为Fe2+催化H2O2产生的·OH,其贡献率占阴极室贡献率的99.61%,阳极室中贡献率最高的是Fe2+催化S2O82−产生的·SO4−,贡献率达49.02%,其次为电子传递氧化途径,贡献率达35.29%,BDD表面产生的·OH贡献率较小,仅11.76%,S2O82−的氧化效果不佳,部分研究中直接忽略不计[13]. 与阳极室降解效果相比,阴极室的Fenton效应体现出显著优势,阴极室的贡献率达阳极室的两倍之多.
-
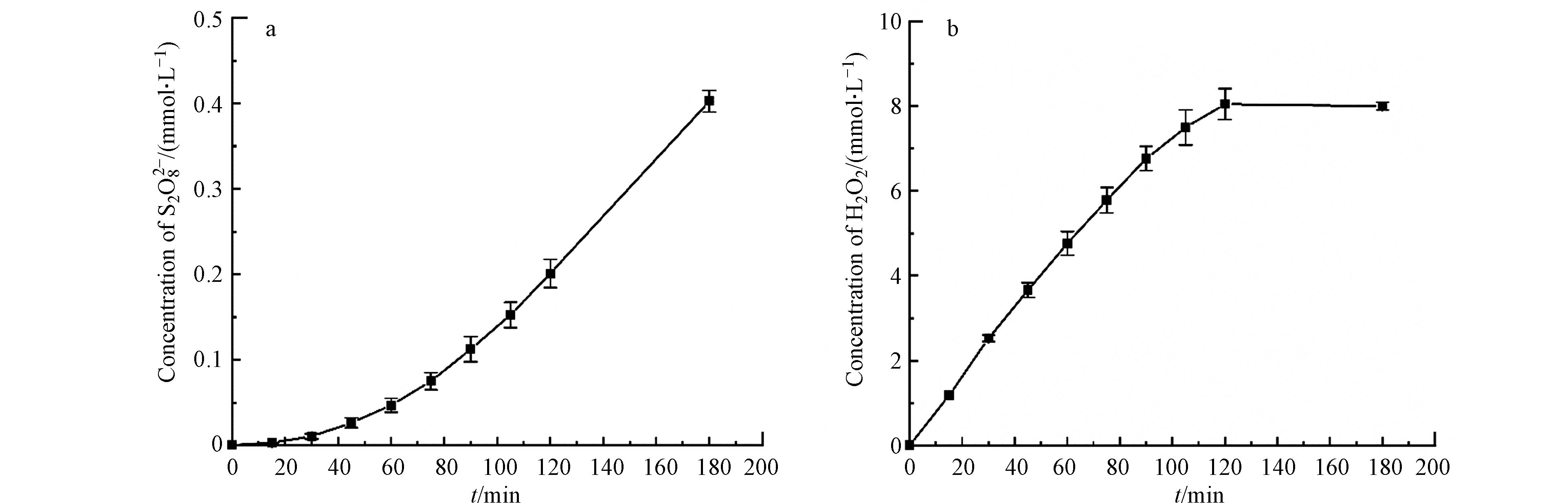
S2O82−和H2O2的浓度可分别通过碘量法和钛盐分光光度法进行测定,通过对这两种氧化性物种随反应时间的积累量测定,可对在加入Fe后被激活产生的·SO4−和·OH的量有大致的了解. 实验中以不添加模式污染物SD的0.1 mol·L−1的Na2SO4溶液为电解液,BDD为阳极,NADE为阴极,外加30 mA·cm−2恒流电流,测定阴极室产生的H2O2量和阳极室产生的S2O82−含量. 图5为180 min内S2O82−和H2O2的浓度变化.
从图5可以看出,阳极室积累的S2O82−浓度在180 min时可达到0.4 mmol·L−1,其本身的氧化能力不强,在加入Fe后S2O82−可以被催化为·SO4−而显示出较好的氧化性. 在酸性条件下,阴极室可通过公式(1)产生H2O2,积累的H2O2浓度在120 min时可达到8 mmol·L−1,180 min时H2O2浓度没有太大变化,这与已有的研究结果相似[19-20],这是因为阴极也会消耗H2O2,如公式(2),导致H2O2积累量逐渐趋于稳定. 随着反应进行H2O2的产量比S2O82−高很多,这也是加入Fe后,阴极室的降解效果比阳极室提升程度更大的原因.
-
在以上双室实验中,控制反应过程中pH保持在3左右,在酸性条件下进行机理探索,但在自然反应条件下,阴、阳极室中pH会因式(3)和(4)而产生变化,因此,在不控制pH的条件下探究了反应过程中的双室pH变化,其结果如图6所示,该结果与李朝林等人研究结果相似[21].
由图6可知,阴极室的pH呈上升趋势,且60 min内pH已呈碱性,约9.69,碱性条件也可以生成H2O2,如公式(5),但其积累量会略有减少[11],其分解如公式(6). 但在pH>5的范围内不利于生成Fe2+,甚至可能会产生的铁离子沉淀,由此不能发挥很好的催化H2O2的性能,因此在不控制pH时,随着反应进行,阴极室的降解效果会受pH升高的影响而降低. 而阳极室的pH仅略有下降,始终保持酸性,对阳极室的降解效果无太大影响. 而在单室反应中,调整初始pH在3左右时,系统内pH不会随反应进行有较大变化,可能是阴阳极产生的OH−和H+相互中和,使系统内酸碱度保持在稳定范围内,因此单室反应中更有利于阴极发挥优异的氧化性能[22].
-
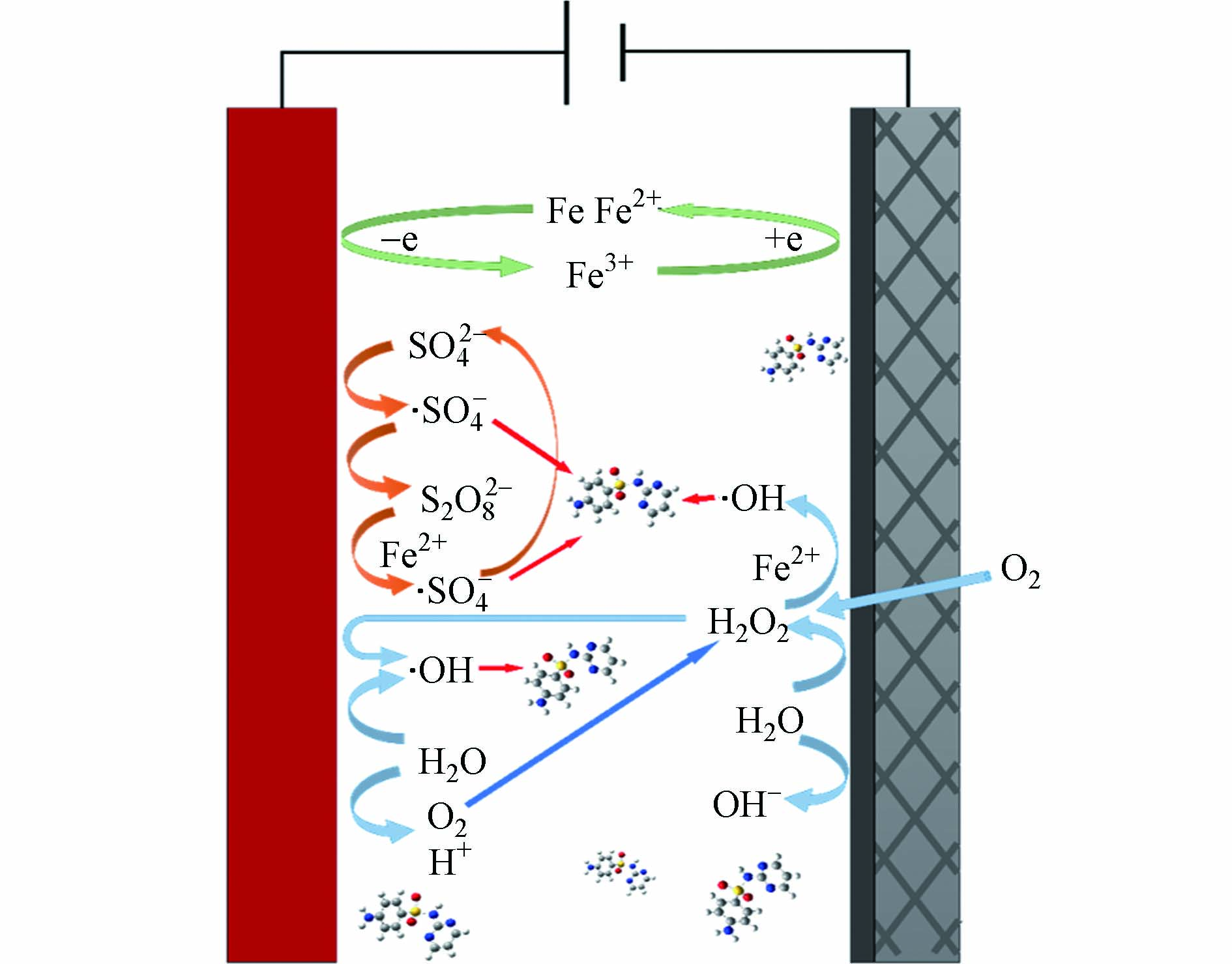
在BDD-Fe-NADE系统中,通过对氧化性粒子的定性测定,部分氧化性粒子的定量测定,推断出如图7示的氧化机理. SO42−在做电解质的同时,可以通过电子转移,少部分通过·OH间接氧化为S2O82−,在Fe2+的催化下产生·SO4−,氧化SD后被还原为SO42−实现循环. 阳极通过电解水产生·OH和O2,·OH可直接氧化SD,O2可转移到阴极上被催化成H2O2. 阴极通过捕获阳极产生的O2和空气中的O2催化产生H2O2,在Fe2+的催化下产生·OH来氧化SD. 而投加的零价Fe可以在酸性条件下被氧化产生Fe2+,而后在催化S2O82−和H2O2的过程失电子变为Fe3+,过量的H2O2可将Fe3+还原为Fe2+,继续参与系统反应,从而实现Fe的循环.
-
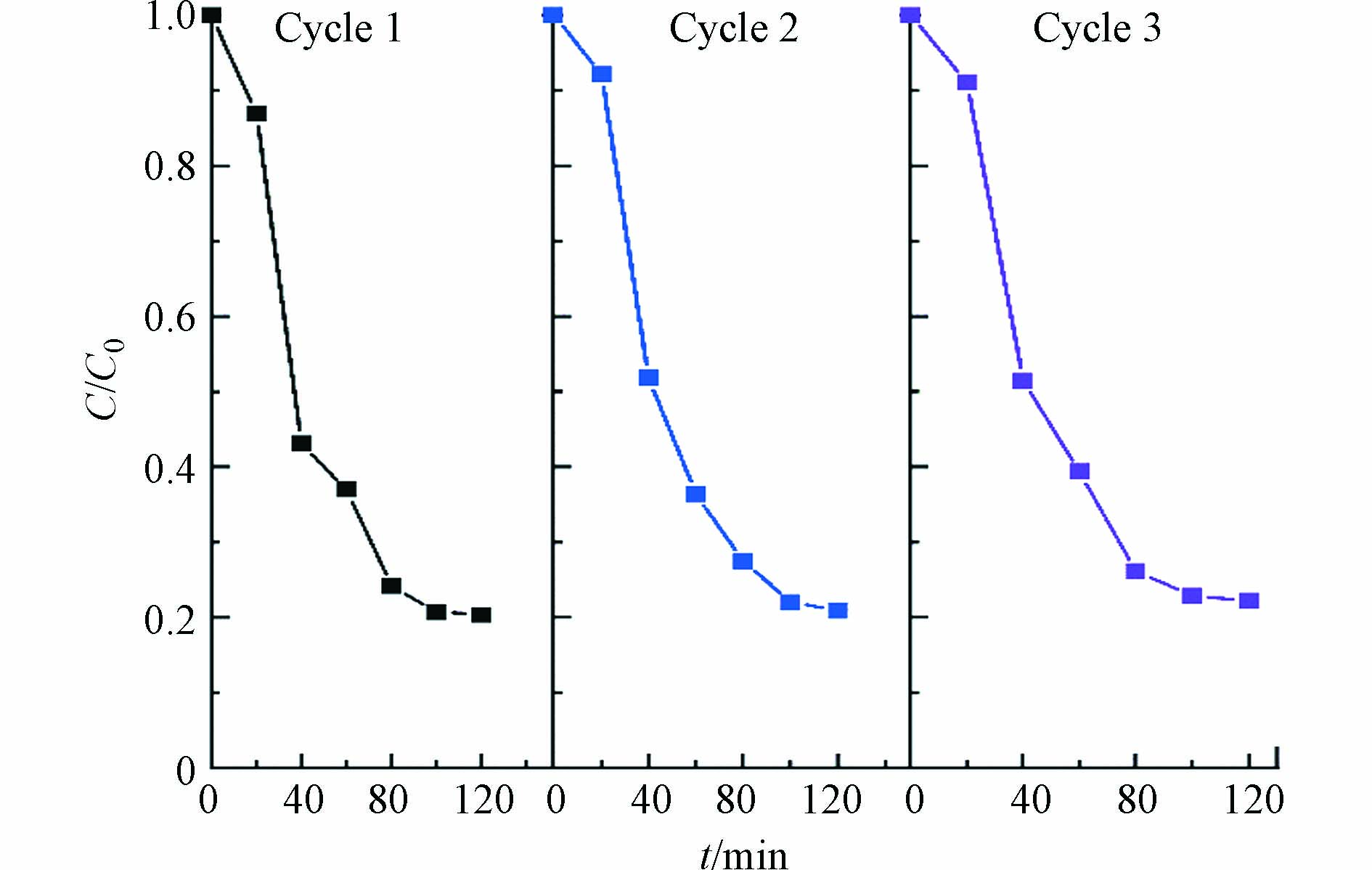
由图8可知,多次循环降解实验后,阴极室降解率仍保持在较为稳定的水平,3次循环测试中,120 min时SD降解率均达到80%,说明本实验体系中的阴极体现了良好的循环性能.
-
使用Gaussian 09软件密度泛函理论(Density Functional Theory, DFT)在B3LYP/6-31+g(d,p)水平下优化SD的几何结构,如图9所示.
福井函数的左导数、右导数和左右导数的平均值分别对应亲电、亲核和自由基反应,即
式中,
$ {\rho }_{\text{N}}\left(r\right)、{\rho }_{\text{N+1}}\left(r\right)、{\rho }_{\text{N-1}}\left(r\right) $ 分别代表体系在原始状态(N电子)、结合一个电子状态(N+1电子)和电离掉一个电子状态(N-1电子)下的电子云密度. 计算$ {\rho }_{\text{N}}\left(r\right)、{\rho }_{\text{N+1}}\left(r\right)、{\rho }_{\text{N-1}}\left(r\right) $ 时,体系的几何结构保持不变.而简缩福井函数(CFF)是将函数收缩到原子上,便于定量比较不同位点上福井函数值的大小. 基于Hirshfeld原子电荷计算简缩福井函数:
基于Hirshfeld原子电荷计算简缩双描述符:
式中,q表示原子电荷数,
$ {{q}}_{{N}}^{{k}}、{{q}}_{{N+1}}^{{k}}、{{q}}_{{N-1}}^{{k}} $ 分别是原子k在原始状态(N电子)、结合一个电子状态(N+1电子)和电离掉一个电子状态(N-1电子)下的电荷数.根据表3的计算结果,可以定量分析SD分子中各原子的福井函数和双描述符大小. 描述亲电反应活性的
$ {\text{f}}_{\text{k}}^{\text{-}} $ 最大正值发生在N10>N25>N2>N1,双描述符$ \Delta {f}_{k} $ 最大负值发生在N25>N10>N2>N1>O11>O12,说明在SD分子中这些位点容易受到亲电试剂攻击. 描述亲核应活性的$ {f}_{k}^{+} $ 最大正值发生在S9>C13>C22>C3>C4,双描述符$ \Delta{f}_{k} $ 最大正值发生在S9>C13>C22,说明在SD分子中这些位点容易受到亲核试剂攻击. 而嘧啶杂环上只有C15呈负电性,且负电性较强,由此推测连接以上原子的化学键较易断裂,但S9的静电荷高达+1.244183,在SD分子中最高,S原子极强的电子缺陷降低了—SO2—基团的平均能,增强了—SO2—基团的稳定性[23]. 下面将通过LC-MS检测结果进行进一步验证与分析. -
通过超高效液相色谱串联高分辨质谱仪对磺胺嘧啶降解过程中中间产物进行测定,测定结果如图10所示. 图10的测定结果表明,磺胺嘧啶在降解过程中产生了m/z分别为225、279、185、171等物质,根据文献查阅和推测,推测出图中显示的6种物质. 除图中显示几种物质外,还检测出m/z为58的小分子物质,推测其为降解过程中产生的乙酸等小物质分子,但因其不作为SD分子降解过程中主要的大分子物质,在此不再重点分析. 结合Gaussian计算中SD分子中的活性位点对其降解途径进行分析,结果如图11所示.
在途径1中,原子N10在Gaussian计算中
$ {f}_{k}^{-} $ 最大正值最大,S9的$ \Delta{f}_{k} $ 最大正值,因此S—N和S—C键容易受到自由基攻击而断裂,—SO2—基团脱离SD分子后,C—N相连后产生同分异构体A或B(m/z=185),该物质也可能继续受到自由基攻击产生物质C和D[24]. 根据苟玺莹等的研究[25],也可能先产生C和D,而后两种物质再相结合产生A和B.在途径2中,N25的
$ \Delta{f}_{k} $ 最大负值最大,因此—NH2容易被氧化为—NO2,产生物质E(m/z=279). E断开N—C键产生物质H(m/z=171),是由于N10拥有较高的福井函数值.在途径3中,由于N1和N2的
$ \Delta{f}_{k} $ 最大负值较大,容易受到亲电试剂攻击,C—N容易断裂生成物质F(m/z=225). 根据杨子增等[25]的研究,推测物质F也可能向物质G(m/z=185)的转化,而后断开N—C键产生物质I(m/z=171). 在分解为J(m/z=93)的过程中,推测物质H先演变为I后,再被氧化脱掉SO42−变成物质J. -
在分析BDD-Fe-NADE系统降解效果和机理实验中,结果表明,BDD-Fe-NADE系统具有优秀的降解效果. 阴极室中Fe2+催化H2O2产生的·OH起主要氧化作用,在阴极室中的贡献率达99.61%;阳极室中,DET和Fe2+催化S2O82−产生的·SO4−起主要氧化作用,在阳极室中的贡献率分别为49.02%和35.29%. 在不添加SD的条件下,阳极室积累的S2O82−浓度在180 min时可达到0.4 mmol·L−1,阴极室积累的H2O2浓度在120 min时可达到8 mmol·L−1. 同时,在初始pH为3时,阴极室pH会随反应进行逐渐增大,阳极室pH显示缓慢的降低趋势. 而在单室反应中,调整初始pH在3左右时,系统内pH不会随反应进行有较大变化,可能是阴阳极产生的OH−和H+相互中和,使系统内酸碱度保持在稳定范围内,因此推测单室反应中更有利于阴极发挥优异的氧化性能. 在SD降解路径分析实验中,利用 Gaussian软件在 B3L YP/6-31+g(d,p)水平优化 SD分子构象,结合密度泛函理论计算电子云密度和福井函数,确定了SD分子中的反应活性位点,利用高效液相色谱串联质谱对中间产物测定,检测到质荷比(m/z)为225、279、185、171等6种中间产物,推测了SD的3种可能降解途径:一是S9—N10和S9—C15断裂,二是N25被氧化,三是N1—C3和N2—C4键断裂. 经探究,BDD-Fe-NADE系统降解抗生素的性能优秀,机理明确,对现今Fe系催化剂激活PS和H2O2降解水中磺胺类抗生素具有重大意义.
BDD-Fe-NADE电芬顿系统降解磺胺类抗生素的作用机理
Degradation mechanism of sulfa antibiotics by H2O2 and PS in BDD-Fe-NADE system
-
摘要: 本研究构建了以BDD电极为阳极,自然空气扩散电极 (natural air diffusion electrode,NADE)为阴极,零价铁作为催化剂的电芬顿系统,深入探索了该系统对磺胺嘧啶(sulfadiazine,SD)的降解效果和机理. 相较于其他系统,BDD-Fe-NADE系统降解优势明显. 为进一步探索系统的作用机理,在BDD阳极和NADE阴极之间加入质子交换膜,构建双室降解系统,分别计算阳极室和阴极室中直接电子转移(direct electron transfer,DET)、H2O2、·OH、S2O82−和·SO4−等氧化性粒子对于磺胺嘧啶降解的贡献率. 结果表明,在阳极室中,电极氧化和Fe2+催化S2O82−产生的·SO4−起主要氧化作用,贡献率分别为49.02%和35.29%;在阴极室中,Fe2+催化H2O2产生的·OH起主要氧化作用,贡献率达99.61%. 在不添加污染物的条件下,阳极室积累的S2O82−浓度在180 min时可达到0.4 mmol·L−1,阴极室积累的H2O2浓度在120 min时可达到8 mmol·L−1. 在初始pH为3时,阴极室pH会随H2O2的生成逐渐增大,阳极室pH由于·OH的生成缓慢降低. 利用 Gaussian软件在 B3LYP/6-31+g(d,p)水平优化 SD分子构象,结合密度泛函理论(Density functional theory,DFT)计算电子云密度和福井函数,确定了SD分子中的反应活性位点,利用高效液相色谱串联质谱(Liquid chromatograph mass spectrometer)对中间产物测定,检测到质荷比(m/z)为 225、279、185、171等6种中间产物,推测了 SD的三种可能降解途径.Abstract: This paper explores the degradation effect and oxidation mechanism of the electro oxidation system with BDD as anode, NADE as cathode and zero valent Fe as catalyst. Compared with other systems, BDD-Fe-NADE system has obvious advantages in degradation. In order to further explore the mechanism of the system, a proton exchange membrane was added between BDD anode and NADE cathode to construct a two-compartment degradation system. The contributions of Direct Electron Transfer (DET), H2O2, ·OH, S2O82−and ·SO4− to sulfadiazine degradation in anode and cathode chambers were calculated. The results show that ·SO4− produced by S2O82− catalyzed by Fe2+ and electrode oxidation play a major role in the anodic chamber, contributing 35.29% and 49.02% respectively. In the cathode chamber, ·OH produced by H2O2 catalyzed by Fe2+ plays a major role in oxidation, contributing 99.61%. In the absence of contaminants, the concentration of S2O82− accumulated can reach 0.4 mmol·L−1 at 180 min in the anode chamber, and the concentration of H2O2 accumulated can reach 8 mmol·L−1 at 120 min in the cathode chamber. When the initial pH is 3, the pH of the cathode chamber gradually increases with the generation of H2O2, while the pH of the anode chamber decreases slowly due to the generation of ·OH. The conformation of SD molecule was optimized at the B3LYP/6-31+g(d, p) level by Gaussian software, and the Fukui function and electron cloud density were calculated with density functional theory (DFT), and the reactive sites in SD molecule were determined. Liquid Chromatograph Mass Spectrometer (LC-MS) was used for the determination of intermediates. The Mass charge ratio (m/z) of intermediates was 225, 279, 317, 185, 171. Three possible degradation pathways were detected finally.
-
Key words:
- BDD anode /
- NADE cathode /
- eletric-fenton /
- hydrogen peroxide /
- persulfate
-

-
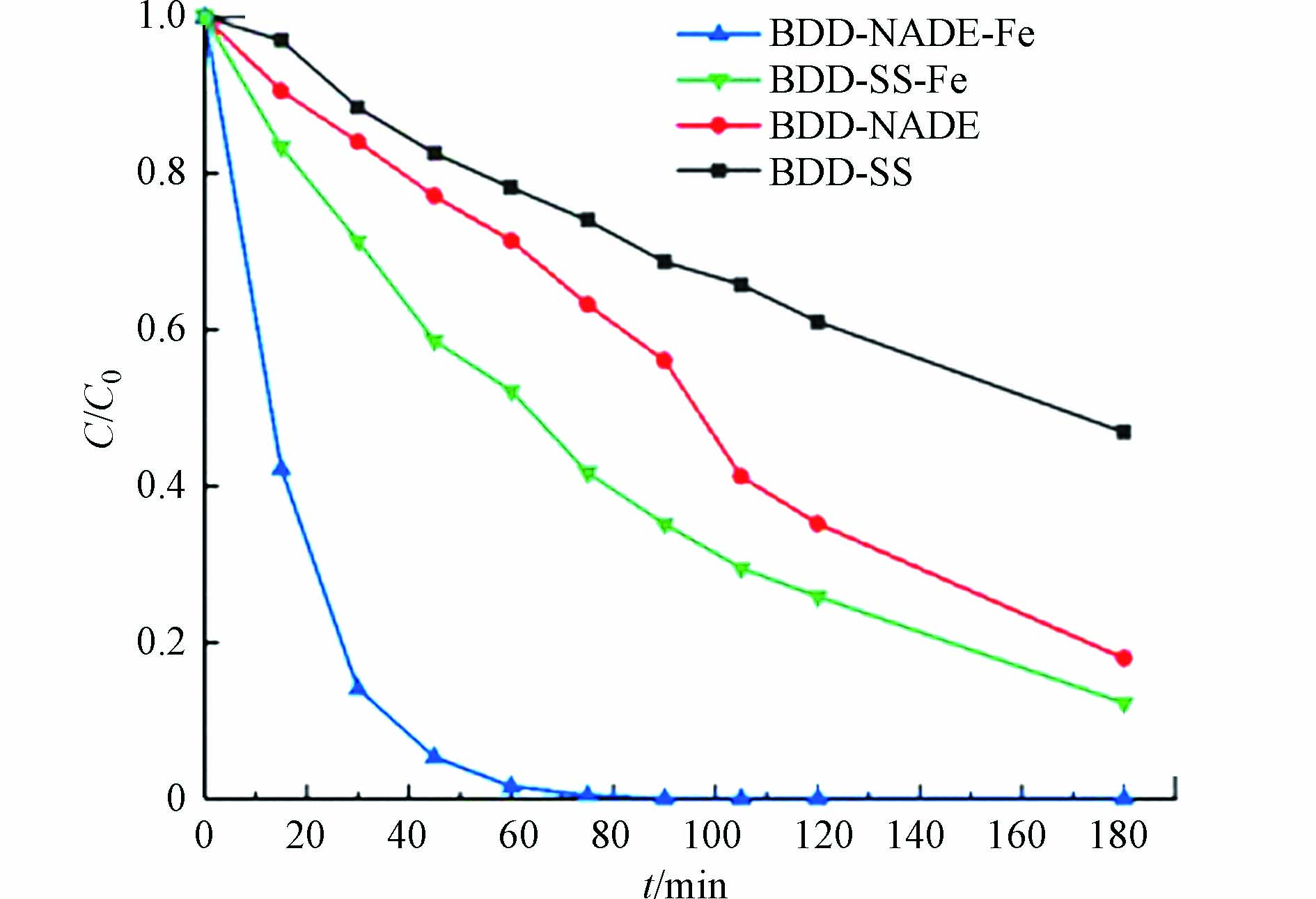
图 2 不同体系对SD的降解效果对比
Figure 2. Comparison of SD degradation effects of different systems
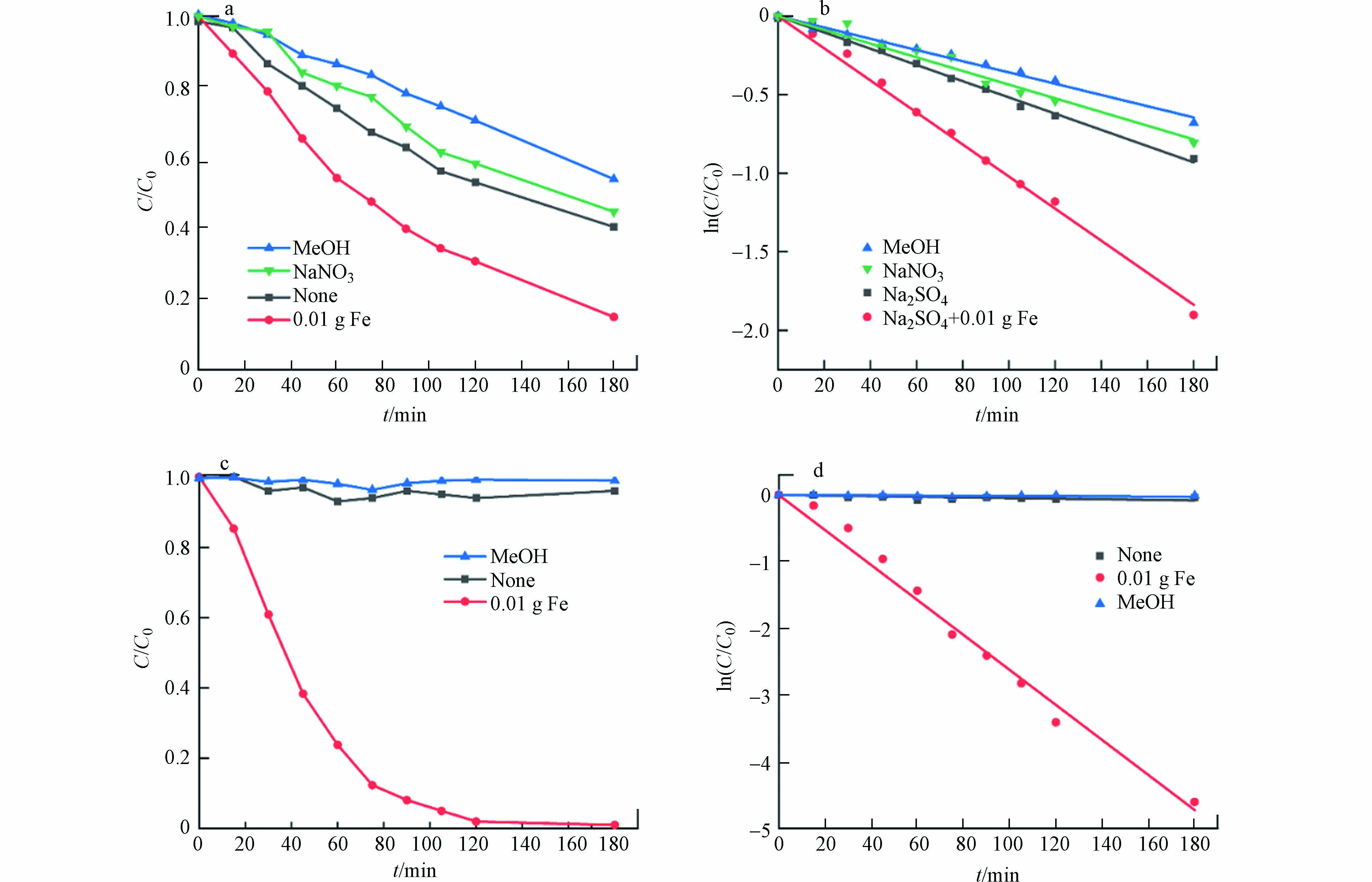
图 3 阳(a、b)、阴(c、d)极室中不同条件下SD的降解效果
Figure 3. Degradation effect of SD in negative and anode chambers under different conditions

图 4 阴、阳极室中主要氧化性物种的相对贡献率
Figure 4. Relative contribution rate of major oxidizing species in anode and cathode chambers

图 6 阴、阳极室反应过程中pH变化
Figure 6. pH changes during the reaction in anode and anode chambers
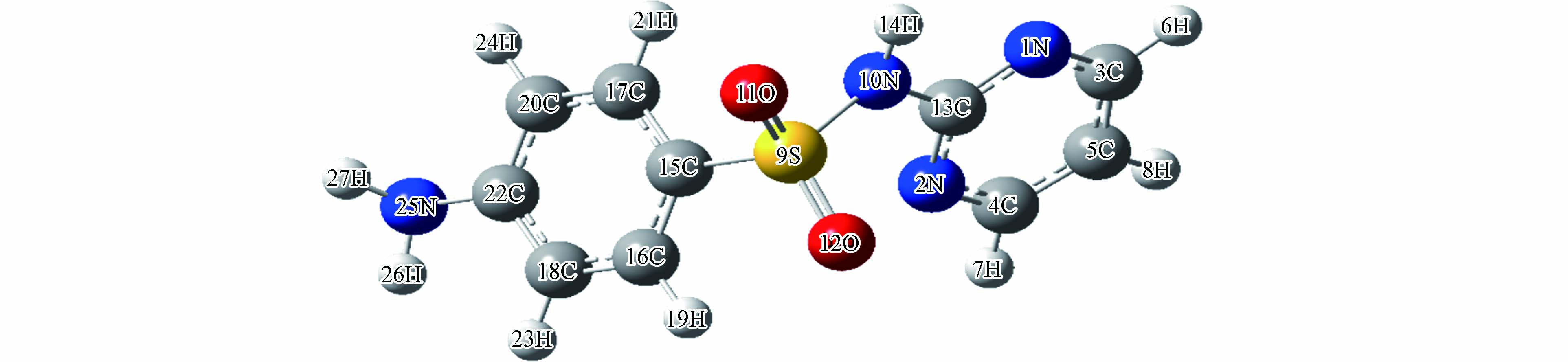
图 9 采用Gaussian在B3LYP/6-31+g(d,p)水平优化后的SD构象
Figure 9. SD conformation optimized by Gaussian at the B3LYP/6-31+ G(d, p)
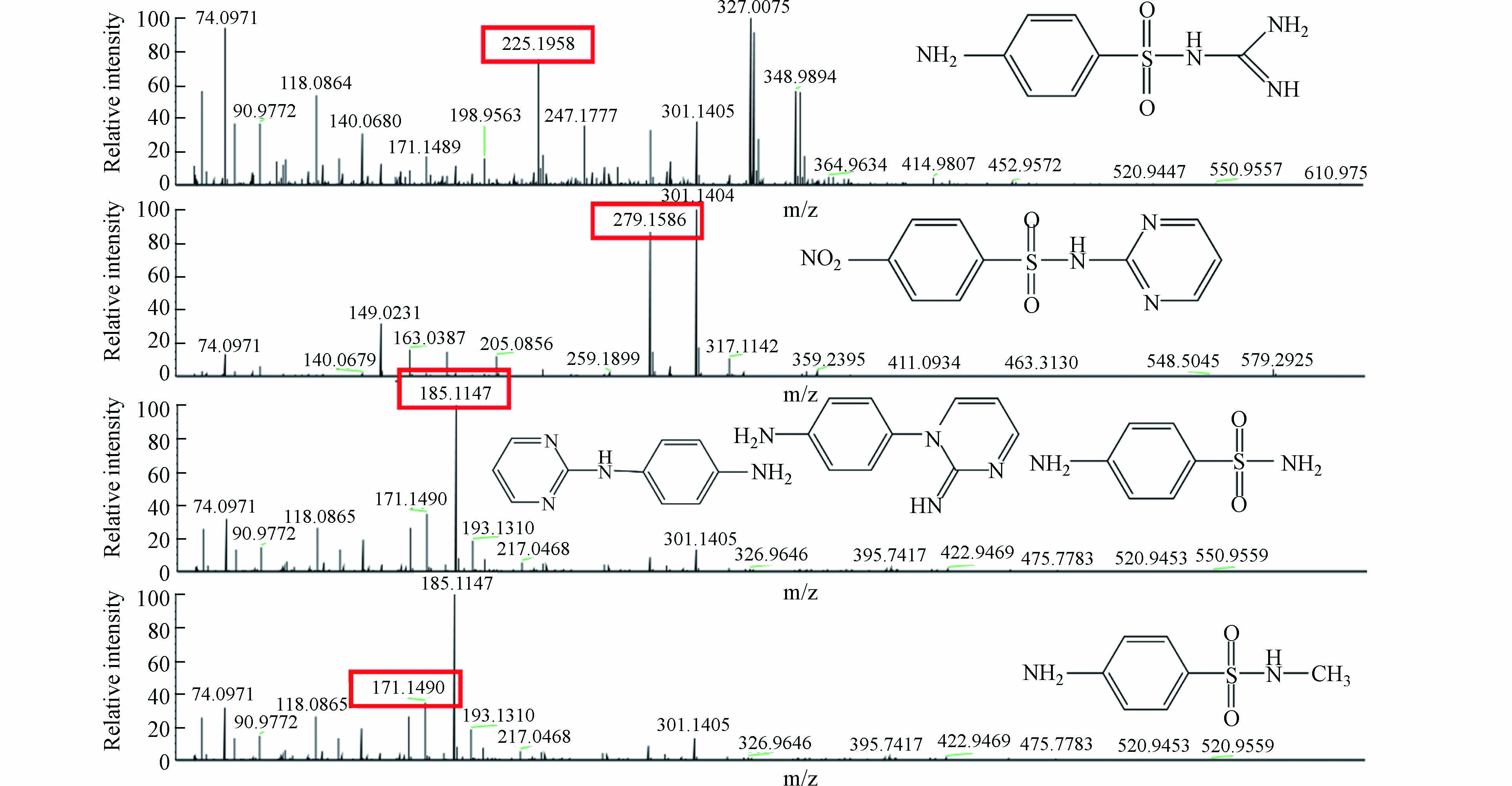
图 10 BDD-Fe-NADE系统中SD中间产物质谱图
Figure 10. Photoelectric degradation of SD intermediate substance in BDD-NADE -Fe system
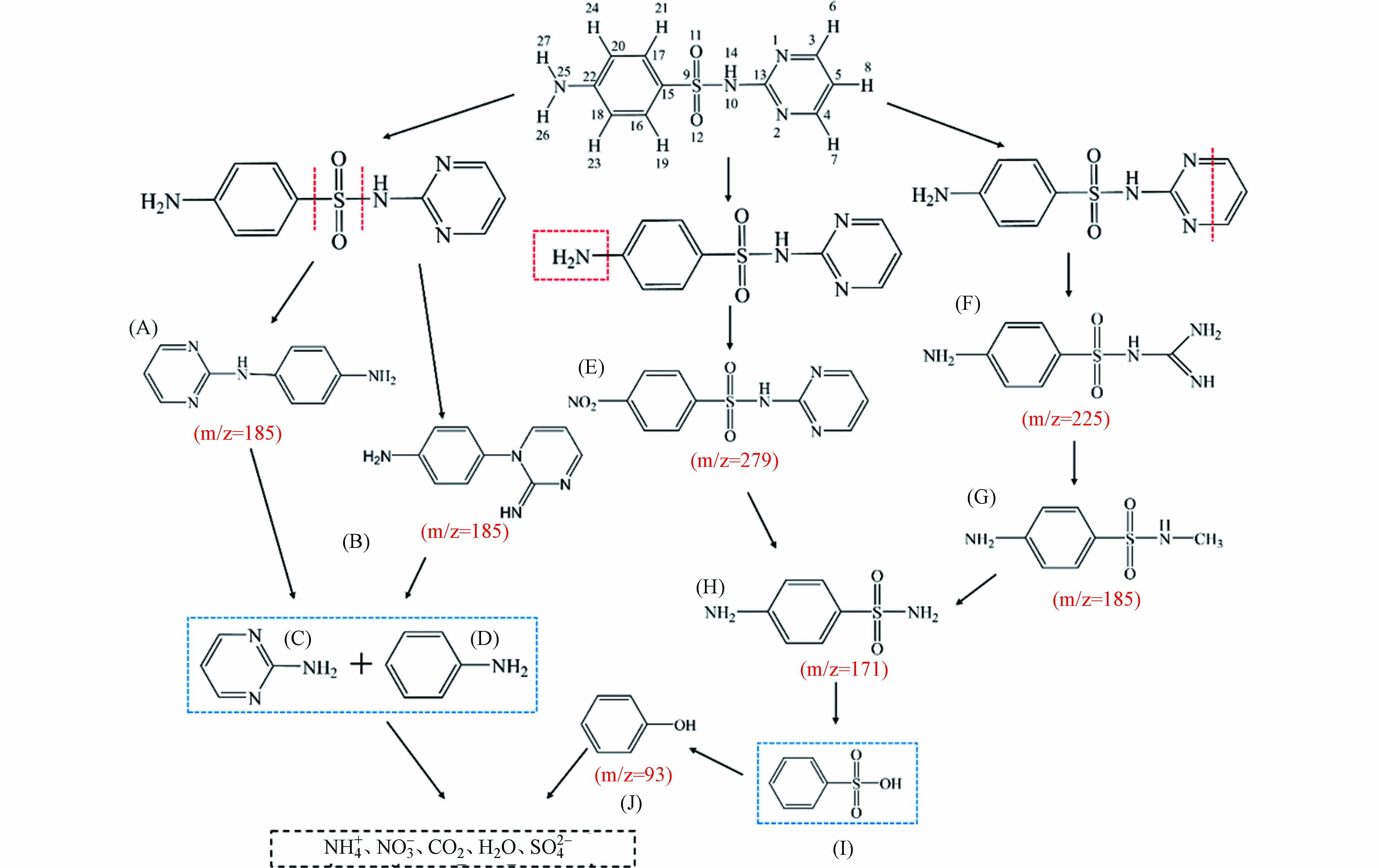
图 11 BDD-Fe-NADE系统中SD降解途径机理图
Figure 11. Mechanism diagram of SD degradation pathway in BDD-NADE -Fe system
表 1 阴阳极室中不同条件降解效果的一级动力学模型
Table 1. First-order kinetic models of degradation effects under different conditions in negative and anode chambers
不同氧化体系
Oxidation system主要氧化性物种
Oxidizing species拟合曲线
Curve相关系数R2
Correlation coefficient R2MeOH (阳极室) DET y=−0.0036x 0.9956 NaNO3 (阳极室) DET、·OH y=−0.0044x 0.9863 Na2SO4 (阳极室) DET、·OH、S2O82− y=−0.0052x 0.9975 0.01gFe (阳极室) DET、·OH、·SO4- y=−0.0102xx 0.9980 MeOH (阴极室) DET y =−1.3312×10−4x 0.4568 None (阴极室) DET、H2O2 y =−4.5303×10−4x 0.7196 0.01 gFe (阴极室) DET、·OH y =−0.0263x 0.9936  下载: 导出CSV
下载: 导出CSV
表 2 阴阳极室中主要氧化性物种的相对贡献率
Table 2. Relative contribution rates of major oxidizing species in negative and anode chambers
氧化性物种
Oxidizing species反应速率/min−1
Reaction rate阳极室贡献率
Anode chamber contribution rate阴极室贡献率
Cathode chamber contribution rate总贡献率
Total contributionDET(阳极室) 0.0036 35.29% — 9.86% DET(阴极室) 1.3312×10−4 — 0.05% 0.04% S2O82−(阳极室) 0.0008 7.84% — 2.19% ·SO4−(阳极室) 0.0050 49.02% — 13.70% ·OH(阳极室) 0.0012 11.76% — 3.29% ·OH(阴极室) 0.0262 — 99.61% 71.77% H2O2(阴极室) 3.1991×10−4 — 0.12% 0.09%
下载: 导出CSV
表 3 SD分子各原子电荷分布及福井函数
Table 3. Atomic charge distribution and Fukui function of SD molecule
序号
Number原子
Atomq(N−1) q(N) q(N+1) $ {{f}}_{{k}}^{{-}} $ $ {{f}}_{{k}}^{{+}} $ $ {f}_{k}° $ $ \Delta {{f}}_{{k}}^{{}} $ 1 N 0.159140 −0.471844 0.036982 0.630984 −0.508826 0.061079 −1.13981 2 N 0.193241 −0.461786 0.032302 0.655027 −0.494088 0.0804695 −1.149115 3 C 0.426275 0.123577 −0.022331 0.302698 0.145908 0.224303 −0.15679 4 C 0.399976 0.124973 −0.014342 0.275003 0.139315 0.207159 −0.135688 5 C −0.158145 −0.124715 0.072764 −0.03343 −0.197479 −0.1154545 −0.164049 6 H −0.024686 0.112320 0.000817 −0.137006 0.111503 −0.0127515 0.248509 7 H −0.023231 0.109609 0.000527 −0.13284 0.109082 −0.011879 0.241922 8 H 0.005523 0.100858 −0.002962 −0.095335 0.10382 0.0042425 0.199155 9 S 0.001378 1.244183 −0.027617 −1.242805 1.2718 0.0144975 2.514605 10 N −0.000527 −0.694635 0.123537 0.694108 −0.818172 −0.062032 −1.51228 11 O −0.000264 −0.549971 0.007608 0.549707 −0.557579 −0.003936 −1.107286 12 O 0.001555 −0.516570 0.025864 0.518125 −0.542434 −0.0121545 −1.060559 13 C −0.051271 0.653111 −0.016108 −0.704382 0.669219 −0.0175815 1.373601 14 H −0.000842 0.309116 −0.003446 −0.309958 0.312562 0.001302 0.62252 15 C −0.004064 −0.199575 0.322091 0.195511 −0.521666 −0.1630775 −0.717177 16 C 0.036056 −0.056802 −0.090401 0.092858 0.033599 0.0632285 −0.059259 17 C 0.007852 −0.079882 −0.092795 0.087734 0.012913 0.0503235 −0.074821 18 C 0.013557 −0.118305 0.159633 0.131862 −0.277938 −0.073038 −0.4098 19 H −0.003718 0.136788 0.002454 −0.140506 0.134334 −0.003086 0.27484 20 C 0.028771 −0.114564 0.167937 0.143335 −0.282501 −0.069583 −0.425836 21 H −0.000556 0.129437 0.002626 −0.129993 0.126811 −0.001591 0.256804 22 C −0.004050 0.299698 0.066359 −0.303748 0.233339 −0.0352045 0.537087 23 H −0.000845 0.083736 −0.006994 −0.084581 0.09073 0.0030745 0.175311 24 H −0.001609 0.085124 −0.007287 −0.086733 0.092411 0.002839 0.179144 25 N −0.000224 −0.655931 0.283023 0.655707 −0.938954 −0.1416235 −1.594661 26 H −0.000013 0.265992 −0.010127 −0.266005 0.276119 0.005057 0.542124 27 H 0.000723 0.266054 −0.010113 −0.265331 0.276167 0.005418 0.541498
下载: 导出CSV
-
[1] HALLING-SØRENSEN B, NIELSEN S N, LANZKY P F, et al. Occurrence, fate and effects of pharmaceutical substances in the environment- A review [J]. Chemosphere, 1998, 36(2): 357-393. doi: 10.1016/S0045-6535(97)00354-8 [2] MARTINEZ J L. Environmental pollution by antibiotics and by antibiotic resistance determinants [J]. Environmental Pollution, 2009, 157(11): 2893-2920. doi: 10.1016/j.envpol.2009.05.051 [3] 高立红, 史亚利, 厉文辉, 等. 抗生素环境行为及其环境效应研究进展 [J]. 环境化学, 2013, 32(9): 1619-1633. GAO L H, SHI Y L, LI W H, et al. Environmental behavior and impacts of antibiotics [J]. Environmental Chemistry, 2013, 32(9): 1619-1633(in Chinese).
[4] 徐维海, 张干, 邹世春, 等. 典型抗生素类药物在城市污水处理厂中的含量水平及其行为特征 [J]. 环境科学, 2007, 28(8): 1779-1783. XU W H, ZHANG G, ZOU S C, et al. Occurrence, distribution and fate of antibiotics in sewage treatment plants [J]. Environmental Science, 2007, 28(8): 1779-1783(in Chinese).
[5] 章强, 辛琦, 朱静敏, 等. 中国主要水域抗生素污染现状及其生态环境效应研究进展 [J]. 环境化学, 2014, 33(7): 1075-1083. doi: 10.7524/j.issn.0254-6108.2014.07.001 ZHANG Q, XIN Q, ZHU J M, et al. The antibiotic contaminations in the main water bodies in China and the associated environmental and human health impacts [J]. Environmental Chemistry, 2014, 33(7): 1075-1083(in Chinese). doi: 10.7524/j.issn.0254-6108.2014.07.001
[6] 叶必雄, 张岚. 环境水体及饮用水中抗生素污染现状及健康影响分析 [J]. 环境与健康杂志, 2015, 32(2): 173-178. YE B X, ZHANG L. Analysis of the pollution status and health risk of antibiotics in water environment and drinking water [J]. Journal of Environment and Health, 2015, 32(2): 173-178(in Chinese).
[7] 刘昔, 王智, 王学雷, 等. 我国典型区域地表水环境中抗生素污染现状及其生态风险评价 [J]. 环境科学, 2019, 40(5): 2094-2100. LIU X, WANG Z, WANG X L, et al. Status of antibiotic contamination and ecological risks assessment of several typical Chinese surface-water environments [J]. Environmental Science, 2019, 40(5): 2094-2100(in Chinese).
[8] 童蕾, 姚林林, 刘慧, 等. 抗生素在地下水系统中的环境行为及生态效应研究进展 [J]. 生态毒理学报, 2016, 11(2): 27-36. TONG L, YAO L L, LIU H, et al. Review on the environmental behavior and ecological effect of antibiotics in groundwater system [J]. Asian Journal of Ecotoxicology, 2016, 11(2): 27-36(in Chinese).
[9] 孙见蕊. 钛基掺硼金刚石薄膜电极的制备、性质及其在废水处理中的应用[D]. 长春: 吉林大学, 2012. SUN J R. Preparation, properties of Ti/BDD electrodes and their applications in wastewater treatment [D]. Changchun: Jilin University, 2012(in Chinese).
[10] PÉREZ J F, LLANOS J, SÁEZ C, et al. Electrochemical jet-cell for the in situ generation of hydrogen peroxide [J]. Electrochemistry Communications, 2016, 71: 65-68. doi: 10.1016/j.elecom.2016.08.007 [11] SUN X P, LV J J, YAN Z H, et al. A three-dimensional gas diffusion electrode without external aeration for producing H2O2 and eliminating amoxicillin using electro-Fenton process [J]. Journal of Environmental Chemical Engineering, 2022, 10(2): 107301. doi: 10.1016/j.jece.2022.107301 [12] 雷阳明, 申哲民, 祝松鹤, 等. 采用气体扩散电极降解酸性红B研究 [J]. 环境化学, 2005, 24(3): 330-333. LEI Y M, SHEN Z M, ZHU S H, et al. Study on degradation of acid red b using gas diffusion electrode [J]. Environmental Chemistry, 2005, 24(3): 330-333(in Chinese).
[13] ZHANG Q Z, ZHOU M H, REN G B, et al. Highly efficient electrosynthesis of hydrogen peroxide on a superhydrophobic three-phase interface by natural air diffusion [J]. Nature Communications, 2020, 11: 1731. doi: 10.1038/s41467-020-15597-y [14] JI Y F, FERRONATO C, SALVADOR A, et al. Degradation of ciprofloxacin and sulfamethoxazole by ferrous-activated persulfate: Implications for remediation of groundwater contaminated by antibiotics [J]. Science of the Total Environment, 2014, 472: 800-808. doi: 10.1016/j.scitotenv.2013.11.008 [15] EPOLD I, DULOVA N. Oxidative degradation of levofloxacin in aqueous solution by S2O82−/Fe2+, S2O82−/H2O2 and S2O82−/OH− processes: A comparative study [J]. Journal of Environmental Chemical Engineering, 2015, 3(2): 1207-1214. doi: 10.1016/j.jece.2015.04.019 [16] 孙智宇. BDD阳极电解硫酸盐制备过硫酸盐的研究[D]. 太原: 太原理工大学, 2020. SUN Z Y. Study on preparation of persulfate by electrolysis of sulfate on BDD anode[D]. Taiyuan: Taiyuan University of Technology, 2020(in Chinese).
[17] DIVYAPRIYA G, NIDHEESH P V. Electrochemically generated sulfate radicals by boron doped diamond and its environmental applications [J]. Current Opinion in Solid State and Materials Science, 2021, 25(3): 100921. doi: 10.1016/j.cossms.2021.100921 [18] NASHAT M, MOSSAD M, EL-ETRIBY H K, et al. Optimization of electrochemical activation of persulfate by BDD electrodes for rapid removal of sulfamethazine [J]. Chemosphere, 2022, 286: 131579. doi: 10.1016/j.chemosphere.2021.131579 [19] FU J L, ZHANG X W, LEI L C. Fe-modified multi-walled carbon nanotube electrode for production of hydrogen peroxide [J]. Acta Physico-Chimica Sinica, 2007, 23(8): 1157-1162. doi: 10.1016/S1872-1508(07)60060-6 [20] ZHOU W, RAJIC L, MENG X X, et al. Efficient H2O2 electrogeneration at graphite felt modified via electrode polarity reversal: Utilization for organic pollutants degradation [J]. Chemical Engineering Journal, 2019, 364: 428-439. doi: 10.1016/j.cej.2019.01.175 [21] LUO H J, LI C L, WU C Q, et al. Electrochemical degradation of phenol by in situ electro-generated and electro-activated hydrogen peroxide using an improved gas diffusion cathode [J]. Electrochimica Acta, 2015, 186: 486-493. doi: 10.1016/j.electacta.2015.10.194 [22] 杨子增. UV/O3降解水中磺胺嘧啶的效能和机理研究[D]. 哈尔滨: 哈尔滨工业大学, 2016. YANG Z Z. Performance and mechanism for degradation of sulfadiazine in water by UV/O3 process[D]. Harbin: Harbin Institute of Technology, 2016(in Chinese).
[23] 余忆玄. 羟基自由基的生成及降解磺胺嘧啶方法和机理[D]. 大连: 大连海事大学, 2019. YU Y X. Mechanism and method of production of hydroxyl radicals and fast degradation of sulfadiazine[D]. Dalian: Dalian Maritime University, 2019(in Chinese).
[24] TENG W, XU J, CUI Y J, et al. Photoelectrocatalytic degradation of sulfadiazine by Ag3PO4/MoS2/TiO2 nanotube array electrode under visible light irradiation [J]. Journal of Electroanalytical Chemistry, 2020, 868: 114178. doi: 10.1016/j.jelechem.2020.114178 [25] 苟玺莹, 张盼月, 钱锋, 等. UV/H2O2降解水中磺胺嘧啶影响因素及机理 [J]. 环境工程学报, 2017, 11(11): 5810-5819. GOU X Y, ZHANG P Y, QIAN F, et al. Influencing factors and mechanism of sulfadiazine degradation by UV/H2O2 [J]. Chinese Journal of Environmental Engineering, 2017, 11(11): 5810-5819(in Chinese).
-
 点击查看大图
点击查看大图
计量
- 文章访问数: 2503
- HTML全文浏览数: 2503
- PDF下载数: 93
- 施引文献: 0











