

-
海洋中的溶解氧对于维持海洋生物的生命活动及海洋生态系统的稳定具有重要作用[1-4]. 海洋溶解氧的收支受物理、生物和化学等过程的共同影响,且这些过程所造成的氧同位素分馏效应是不同的(图1)[5-8]. 海洋溶解氧的主要来源为海洋表层的大气溶解氧和浮游植物光合作用产氧. 当混合层中的氧气达到溶解平衡后,其氧同位素组成(δ18O)大约为+0.7‰,而光合作用产生的氧气其δ18O与源海水相同,即δ18O约为-23.5‰[7]. 大洋混合层中溶解氧的δ18O为二者的混合值,δ18O越低指征该海域生产力越高[9]. 河口或受冰川融水影响的海域海水的δ18O值偏低,如受长江冲淡水影响的长江口及其毗邻海域[10]. 当夏季海洋表层的生产力较高时,长江口海洋溶解氧的δ18O值可能会低至-5‰[11]. 氧气的消耗过程主要为生物呼吸作用耗氧. 由于生物在呼吸过程中倾向于消耗较轻的氧同位素(16O),使得水体中剩余的溶解氧更富集较重的氧同位素(18O). 因此,随着呼吸作用的进行,海洋溶解氧浓度降低,且剩余溶解氧的δ18O升高. 海洋中的呼吸耗氧遵循封闭系统瑞利分馏的原则,与氧浓度呈现非线性的变化关系(图1)[12]. 不管是在开阔大洋还是在河口海岸的陆架区域,生物呼吸作用不仅能发生在水柱中(water column respiration, WCR),也发生在底层沉积物中(sediment oxygen respiration, SOR)[13],且这两种呼吸作用显示出不同的氧同位素分馏效应(图1). 观测到的海洋浮游生物WCR氧同位素分馏系数范围为-26‰至-14‰[14],而培养实验得到的SOR氧同位素分馏系数为-5‰至-1‰[15]. 因此,两种呼吸过程会造成迥异的溶解氧δ18O演化趋势,且普遍认为SOR对于水体中溶解氧的氧同位素分馏效应较小. 利用这个特征,前人区分及量化分析了海洋耗氧过程中水柱呼吸与沉积物呼吸的占比[11,16]. 另外,洋流导致的物理输运,如平流、扩散等,也会影响海洋溶解氧浓度和同位素组成. 但物理作用引起的水团混合对δ18O和氧浓度的影响是线性的,与呼吸作用不同(图1). 基于溶解氧同位素在氧循环各过程中的特性,尤其是在不同耗氧过程中分馏效应的差异,使得其成为研究海洋溶解氧收支平衡的重要指标之一[6].
由于人类活动的影响,河口海岸区域的营养盐输入激增,导致长江口及其周边海域富营养化,引发海洋缺氧事件,且近年来观测到的缺氧区面积也在逐渐扩大[17-20]. 长江口缺氧区的形成机制复杂,海水的层化、海底地形及富营养化引起的耗氧过程等都会加剧底层海水的缺氧程度[21-24]. 由于长江口海域的整体水深较浅,底层沉积物中富含海洋自生及陆源输入的有机质[25],底层水受沉积物再悬浮的影响也会造成额外的有机质分解耗氧[26]. 因此,生物通过呼吸作用降解有机物消耗氧气的过程不仅发生在水柱中,沉积物中的生物呼吸也是不容忽视的过程[27]. 探究WCR和SOR对长江口缺氧的贡献,对于探讨该海域缺氧区的形成机制具有重要意义.
测量海洋溶解氧的同位素组成需要较为严格的采样、提取和纯化条件,具有一定的难度. 利用溶解氧同位素探究海洋氧循环的研究多集中于美国东西岸及墨西哥湾等海域[28-30],并没有在各大海域得到广泛的应用.
本文详细描述了海洋溶解氧的采集、实验室提取和纯化及呼吸作用暗培养的方法,并阐述了氧同位素的质谱仪测量和数据校正的具体过程. 同时,利用2020年于长江口采集的混合层海水和底层沉积物,分别进行了暗培养实验,确定了长江口特定的WCR和SOR的氧同位素分馏系数,并估算了水柱呼吸和沉积物呼吸在总耗氧量中的占比,分析了长江口缺氧区占主导的耗氧机制.
-
自然界中氧的稳定同位素有16O、17O、18O,丰度分别为99.76%、0.04%和0.20%[31]. 氧同位素组成及氧气氩气之间的气体比值通常用相对丰度δ值(‰)来表示.
其中,18R = 18O/16O,即18O的丰度与16O丰度的比值,O2/Ar指的是气体中氧气与氩气的含量比,下标sam和std分别代表样品和标样,δ值的单位为千分比(‰). 本研究以空气为标样,即所有样品的δ18O和δO2/Ar值都是相对于空气相应值的偏差. 因此根据定义空气本身的δ18O和δO2/Ar值为0‰.
微生物在呼吸作用中倾向于先消耗轻的氧同位素(16O),使剩余的溶解氧中富集较重的氧同位素(18O). 呼吸作用过程中造成的氧同位素分馏程度用分馏系数α或ε来表示 [12,32],定义如下:
其中,respired代表生物呼吸作用消耗的氧气,residual代表剩余的氧气. 呼吸作用的α值通常为略小于1的数值. 对于海洋生物来说,其观测的范围为0.974–0.986,对应的ε值为-26‰至-14‰[12,14],ε是α的千分比形式(‰). 海洋生物的呼吸作用遵循封闭系统瑞利分馏的原则,即[33]:
其中,residual代表呼吸作用之后剩余的溶解氧,initial代表初始的溶解氧,f是呼吸作用后剩余溶解氧占初始溶解氧的比例,即
$ \left[{O}_{2}\right]/{\left[{O}_{2}\right]}_{0} $ . 由公式(5)可得,随着溶解氧被生物呼吸作用消耗,剩余溶解氧的δ18O逐渐升高,且与氧浓度呈非线性变化,其中的演化关系与分馏系数α相关. 若耗氧过程中的α为未知量,在公式(5)两边取对数后可变换为:结合公式(1)和(4)及
$ {10}^{3}\mathrm{l}\mathrm{n}(1+X)\approx X $ 的近似关系,公式(6)可变换为:通过观测呼吸作用过程中不同时间节点的氧浓度和δ18O值,利用δ18O和
$ \mathrm{l}\mathrm{n}\left(\text{[}{\text{O}}_{\text{2}}\text{]}/{\text{[}{\text{O}}_{\text{2}}\text{]}}_{\text{0}}\right) $ 作图,即可通过二者间的斜率确认呼吸作用过程的氧同位素分馏系数ε. -
大气中氧气的δ18O(0‰)与溶解氧的δ18O截然不同,因此溶解氧采集过程中需要严格防止大气污染. 为保证采集过程中的真空条件,制作了带有真空阀门(LouwersHanique®)的容积为500 mL的样品瓶(图2a). 采样前,清洗过后的样品瓶需根据采集海水的量加入氯化汞(HgCl2)饱和溶液. 最终采集的海水样品中氯化汞浓度需大于2×10−5 kg∙L−1,以确保氯化汞阻断所有生命活动,达到固定海水溶解氧的目的. 加完氯化汞的样品瓶需在低温(< 40°C)、通风的条件下缓慢烘干,待饱和氯化汞溶液凝结为晶体后,再将样品瓶移至实验室真空管线上抽真空(图3). 当样品瓶中的压力小于10−3 Pa后,关闭瓶上方的真空阀门,并在阀门支管中注入蒸馏水、盖上橡胶帽. 阀门支管内的水可辅助真空阀门的O型圈保持样品瓶的真空度. 样品瓶在取样前后都应将阀门支管内注满水. 如此,样品瓶的真空度最长可保持6个月左右.
采集不同深度的原位海水需借助于船载的温度盐度探测仪及海水收集装置(Sea-bird Costal Hydrocat, CTD). 为防止大气污染,海水溶解氧样品需第一个于CTD上采样. 利用橡胶软管将CTD中的水引入样品瓶的阀门支管内,冲洗5–10 s,直至支管内原先注满的蒸馏水被替换为海水. 之后,在保证支管内有水溢出的状态下缓慢打开真空阀门至最下方的O型圈解除密封,海水样品会沿阀门下方的管壁进入真空瓶内. 此时可以观察到大量的气泡从海水中溢出,进入到样品瓶的顶空中. 在质谱仪上测量溶解氧δ18O需约2×10−5 mol O2,因此一般需采集约100 mL的海水. 但对于缺氧区等溶解氧含量极低的海水,可根据其溶解氧浓度适当增加采集的海水量,确保溶解气体中的O2达到质谱仪的检测限. 但总的来说,采集的海水体积最好不超过样品瓶体积的50%,此时残留在溶解态中的气体占比小于2%,对顶空中气态氧气δ18O值的影响可以忽略. 如果样品瓶顶空体积过小,导致其气压过高,将会阻碍溶解气体从海水中的释放. 此时,需要根据N2、O2、Ar的溶解度和分压对气态和溶解态的气体同位素组成及气体比值进行校正.
-
为确定WCR和SOR的氧同位素分馏系数,本研究在实验室中利用海水和海洋沉积物进行了生物呼吸作用暗培养实验. 依托2020年夏季长江口共享航次(NORC2020-03),本研究于“浙渔科2”号科考船上通过CTD和箱式采泥器分别采集了原位的混合层海水和底层沉积物(122.56°E,29.4472°N)用于实验室暗培养.
-
根据公式(7)可知,计算呼吸作用分馏系数ε需要初始、及不同时间节点的氧浓度和δ18O值,以得到δ18O–
$\text{ln}\left(\text{[}{\text{O}}_{\text{2}}\text{]}/{\text{[}{\text{O}}_{\text{2}}\text{]}}_{\text{0}}\right)$ 的斜率. 培养实验的初始样品(t = 0)即为原位海水,经CTD直接采集混合层海水至提前准备好的图2a的真空样品瓶中. 接着,需进行培养的海水会由CTD采集至图2b所示的带磨口玻璃塞的、提前用锡纸包裹住瓶身的溶解氧瓶(250 mL)中. 待溶解氧瓶被注满后,塞上磨口玻璃塞并确保槽口中留有一定的海水用于液封. 随后用封口膜(Parafilm)包裹住槽口防止液封的水在运输的过程中从溢出. 取样后的溶解氧瓶必须全部以不透光的锡纸包裹,确保实验在无光的条件下进行,防止光照引起光合作用. 当达到设定的培养时间时,需将溶解氧瓶中的海水取样至真空样品瓶中固定保存,供后续同位素测量. 具体操作过程为,将槽口中液封的水倒掉后取下磨口玻璃塞. 然后将橡胶软管插至瓶底,利用洗耳球把瓶内的海水吸出并利用虹吸作用形成固定的水流. 之后将橡胶软管迅速置于真空样品瓶的阀门支管内,在确保支管有水流溢出的情况下缓慢打开阀门,培养后的海水样品随即被吸入加有氯化汞的真空瓶内固定保存.为获得准确度较高的δ18O–
$ \mathrm{l}\mathrm{n}\left(\text{[}{\text{O}}_{\text{2}}\text{]}/{\text{[}{\text{O}}_{\text{2}}\text{]}}_{\text{0}}\right) $ 斜率,通过培养实验获得的海水最好能有较大范围的氧同位素和氧浓度变化. 因此,本研究于航次中除初始样品(t = 0的原位海水)外,采集了3个同站位同深度的混合层海水样品,分别于培养的第16天、第23天、第32天取样至真空瓶内固定保存,供后续的同位素测量. -
为得到纯粹的沉积物呼吸作用分馏系数,排除海水中微生物的影响,沉积物培养的溶液为实验室配制的人工海水. 人工海水的盐度是35.00‰,其组成为每升Milli-Q水中加入35g NaCl和0.5 g NaHCO3. 配制后的人工海水需加入磁力转子,在磁力搅拌器的作用下与大气平衡约24 h,使人工海水中的溶解气体与大气达到溶解平衡.
沉积物培养实验的流程与海水培养实验大致相同,具体操作为:于溶解氧瓶中加入5 g沉积物,再加入溶解平衡后的人工海水并注满,塞上磨口玻璃塞并液封保存. 当培养达到目标时间后将水样取至真空瓶中保存,供后续同位素测量. 需特别说明的是,沉积物培养实验的t = 0样品为达到溶解平衡的人工海水,此时的溶解气体未受到任何沉积物呼吸的影响,可视作溶解氧的初始状态. 由于沉积物中的生物含量及有机质含量较高,因此沉积物培养实验的呼吸速率比海水培养的呼吸速率快. 沉积物呼吸暗培养的水样分别在培养开始后的第1天、第2天、第3天取样完成.
-
溶解氧的提取与纯化方法参照了气体溶解平衡取样法[34]. 首先,取样完成后的真空样品瓶需在摇床上(2 r·s−1)摇晃至少48 h,使顶空中的气体与残留在液态中的溶解气体达到同位素平衡. 之后,将样品瓶连接到图3a所示的溶解氧提取线路上. 打开真空泵(Pfeiffer Vacuum Duo 3),抽真空5 min左右. 然后缓慢打开样品瓶上的阀门,样品瓶中的水会在内外压力差的作用下被排出到融水收集瓶内. 注意,排水至最终阶段时需留存约1 mL的液体没过阀门最下端的O型圈,此时快速关闭阀门,以防止瓶内的气体被真空泵抽走.
排水后的样品瓶内即为需要纯化和收集的溶解气体. 将样品瓶连接到溶解氧纯化线路上(图3b),瓶身底部没入-30°C的乙醇浴中,将剩余在样品瓶中的水全部冻结. 在真空管线的另一端接入样品收集阀,收集阀中填充有干燥并去气的硅胶颗粒(45–60 mesh, Sigma). 打开真空管线上所有的阀门使样品瓶与真空泵之间形成通路并抽真空,最终管线的真空度需达到10−3 Pa. 之后,关闭图3b中的V1, V2阀门,使管线与真空泵隔绝,并将两个冷肼及样品收集阀都没入到液氮(-196°C)中. 此时打开样品瓶上方的真空阀门,使溶解气体扩散至真空管线内. 溶解气体中的水蒸气、二氧化碳会经由两个液氮冷肼去除,纯化后的气体为氧气、氮气和氩气,最终被收集于样品阀中的硅胶颗粒上,整个过程持续约45 min.
-
溶解气体的同位素及气体比值是在双路进样的气体同位素质谱仪(Thermo Fisher Delta V Plus)上进行的. 将样品收集阀连接到质谱仪双路进样系统的样品气缸侧,加热硅胶10min使气体充分混合. 之后打开样品收集阀,纯化后的氧气、氮气、氩气即扩散进入质谱仪的样品气缸. 参考气侧为氮气、氧气、氩气占比(N2:O2:Ar)分别为63.93%、34.35%、1.72%的混合气体(纯度均为99.999%). 该气体比值与平衡溶解气体的N2:O2:Ar比值接近,会减小因参考气与样品之间气体比值不同而对氧同位素值造成的质量干扰. 开始同位素测量后,设置质谱仪的测定时间为26 s, 循环15次为一次测量,共测定3次,总耗时约45 min. 质谱仪配备有质量数为28、29、 32、 34、40的法拉第杯,因此可同时测定溶解气体的δ15N、δ18O、δO2/Ar的组成. 由同位素质谱仪得到的初始数据为相对于参考气的同位素及气体比值的数据(
$ {\delta }_{\text{vs.ref}} $ ).高精度高准确度的同位素及气体比值数据需遵循严格的同位素校正流程. 具体需要四个步骤:零点校正、质量干扰校正、空气标样校正和第二标样对比验证[35].
(1)零点校正(zero enrichment):在样品气缸和参考气气缸中都引入参考气,对比测量. 若两侧的气缸及毛细管流速都能够在长时间测量中保持平衡,且没有明显的偏移,则最终零点校正的δ18O, δO2/Ar平均值都应接近于0. 在2020.06 —2020.10和2020.10—2021.03两个测量周期(每根离子源灯丝的寿命期为一个测量周期)共做了79次零点校正测量,观测到的δ18O, δO2/Ar均接近于0(表1). 之后,将所有样品的δ18O和δO2/Ar值都减掉零点的δ18O和δO2/Ar平均值,即完成零点校正. 如果长时间、多次观测到偏离于零点较大的δ18O, δO2/Ar值,应及时检查仪器状态是否正常.
(2)质量干扰校正(mass interference correction or “chemical slope” correction):由于样品的N2:O2:Ar比会随着氧气组分的不同而改变,因此会与参考气的N2:O2:Ar产生差异. 样品与参考气气体组分的差异会引起质量干扰(mass interference),观测的结果表明氮气越多,实测的δ18O值越低(表1). 因此,在每个测量周期中,都应该首先确定质量干扰的斜率. 具体操作方式为,两侧都加入参考气,并在样品气缸中少量多次加入高纯氮气,改变样品侧的N2:O2:Ar比值,并测定每个节点δΝ2/O2值对应的δ18O和δO2/Ar值,即可分别得到δ18O – δΝ2/O2和δO2/Ar – δΝ2/O2的质量干扰斜率(m). 校正公式为:
其中
$ {\delta }_{\mathrm{M}\mathrm{I}-\mathrm{c}\mathrm{o}\mathrm{r}\mathrm{r}} $ 和$ {\delta }_{\mathrm{Z}\mathrm{E}-\mathrm{c}\mathrm{o}\mathrm{r}\mathrm{r}} $ 分别为质量干扰校正后和零点校正后的δ18O或δO2/Ar值. 在两个不同的测量周期,质量干扰斜率相差接近两个数量级,这是因为质谱仪每次更换灯丝后的线性(linearity)及灵敏度(sensitivity)都会发生变化. 经验表明,质谱仪的线性斜率越高,质谱仪的灵敏度会提高,但质量干扰的斜率也会升高. 因此,在样品气体量足够的情况下,可适当牺牲仪器的灵敏度,防止质量干扰的斜率过高,有助于提高同位素数据的准确性.(3)空气标样校正(correction versus air standards):由于各实验室所用的参考气组成各不相同,因此同位素及气体比值数据需相对于同期测定的国际标样空气的数据进行标准化. 即,将公式(1), (2)中的分母由参考气的18R值替换为空气的18R值. 由于空气的存留时间远大于其大气混合时间,因此全球大气是混合均匀的,其氧同位素及气体比值组成也具有全球均一性. 在本研究测样过程中,每天都会于实验室外采集空气,在真空管线上干燥、纯化,并在质谱仪上测量其δ18O和δO2/Ar值. 在2020.06 —020.10和2020.10—2021.03两个测量周期共分别测量了60次和54次空气标样,δ18O和δO2/Ar的测量精度如表1所示,均与国际上同等的气体氧同位素实验室的精度达到一致[36-38]. 观测到的空气标样精度即为本实验室同位素测量的外部精度(external precision). 之后,需要将质谱仪上得到的相对于参考气的样品数据转换为相对于空气标样的数据,公式为:
其中
$ {\delta }_{\text{MI-corr}} $ 为零点校正和质量干扰校正后的,相对于参考气的δ18O或δO2/Ar值,$ {\delta }_{\text{air}} $ 为测量周期所有空气数据相对于参考气的平均值(同样需进行零点校正和质量干扰校正). 最终得到的相对于空气的$ {\delta }_{\text{vs.air}} $ 即为最终所需的样品的δ18O或δO2/Ar值数据.(4)第二标样对比验证:本研究以实验室制作的平衡溶解气体为第二标样,验证了溶解氧提取、纯化方法及同位素矫正流程的准确性. 取1 L 去离子水,加入400 μL饱和氯化汞溶液阻断生命活动. 在磁力搅拌器的作用下使溶解气与大气在室温(22°C)平衡至少24 h,注意磁力搅拌器转速不能过快,以防造成溶液的过饱和状态. 后续取样和提取纯化过程与“1.3”和“1.4”中描述的类似,即利用软管和虹吸作用将水取至真空样品瓶中,并于真空管线上完成提取和纯化,最终于质谱仪上测量. 基于本方法测得的平衡溶解气的δ18O和δO2/Ar值如表1所示,与22 °C下的理论值0.73‰和-93‰接近[39-41],证明本研究所述的溶解氧同位素及气体比值的测定方法是准确可靠的.
-
通过对长江口采集的混合层海水和底层沉积物进行呼吸作用暗培养,分别测定了长江口水柱呼吸作用(WCR)和沉积物呼吸作用(SOR)的氧同位素分馏系数
$ {\epsilon }_{\text{WCR}} $ 和$ {\epsilon }_{\text{SOR}} $ . 根据公式(7)可知,δ18O–$ \mathrm{l}\mathrm{n}\left(\text{[}{\text{O}}_{\text{2}}\text{]}/{\text{[}{\text{O}}_{\text{2}}\text{]}}_{\text{0}}\right) $ 之间的斜率即为ε. 通过对不同培养时长的溶解氧瓶取样,本研究获得了从呼吸作用尚未开始(t0样品),到溶解氧消耗至约50%的一系列样品,并测量了其δ18O和δO2/Ar组成(表2).各时间节点的溶解氧浓度可通过对应的δO2/Ar值计算得到. 氩气与氧气的物理性质较为相似,比如溶解度、扩散速率、对温度的敏感度等[42]. 但是Ar是惰性气体,海洋中Ar的含量主要受物理因素如压力变化、海水温度变化和气泡注入等影响,而不受生物过程的影响[43]. 在整个培养实验过程中,温盐条件已知,Ar浓度保持不变,即为其饱和溶解度. 因此,可以通过海水的温度和盐度,精确计算出Ar的浓度,再根据O2/Ar比值,计算得到海水中氧气的浓度. Ar浓度(μmol∙kg−1)计算公式为[39]:
其中,
$ {T}_{S}=\mathrm{l}\mathrm{n}\left(\dfrac{298.15-t}{273.15+t}\right) $ ,t代表温度(℃),S代表盐度(‰, PSS),${\mathrm{A}}_\mathrm{0}$ = 2.79150,$ {\mathrm{A}}_{1} $ = 3.17609,$ {\mathrm{A}}_{2} $ = 4.131116,$ {\mathrm{A}}_{3} $ = 4.90379,$ {\mathrm{B}}_{0} $ = −6.96233×10−3,$ {\mathrm{B}}_{1} $ = −7.66670×10−3,$ {\mathrm{B}}_{2} $ = −1.16888×10−2. 本实验的培养实验温度为22℃,混合层海水的盐度为27.84‰,沉积物培养的人工海水盐度为35.00‰. 由氩气浓度、公式(2)及大气中的O2/Ar比值(0.21:0.00934),即可计算溶解氧的浓度(μmol∙kg−1)为:最后,通过对δ18O–
$ ln\left(\text{[}{\text{O}}_{\text{2}}\text{]}/{\text{[}{\text{O}}_{\text{2}}\text{]}}_{\text{0}}\right) $ 作图,其斜率即为分馏系数ε(图4). 基于本实验,测定出长江口水柱中生物呼吸的分馏系数$ {\epsilon }_{\text{WCR}} $ 为-20.9‰,沉积物呼吸的分馏系数$ {\epsilon }_{\text{SOR}} $ 为-8.8‰. 对应的分馏系数$ {\alpha }_{\text{WCR}} $ 和$ {\alpha }_{\text{SOR}} $ 分别为0.9791和0.9912. 水柱呼吸和沉积物呼吸本质上都是微生物通过分解有机质消耗氧气,但是二者对溶解氧的消耗所产生的氧同位素分馏效应却有约12‰的明显差异. 这是由于沉积物呼吸耗氧时,氧同位素的分馏不仅由生物呼吸作用引起,氧气在沉积物中的扩散也会造成同位素分馏. 扩散作用的氧同位素分馏系数为0.9978[41],远高于不受扩散作用影响的水柱呼吸作用的$ {\alpha }_{\text{WCR}} $ 值. 基于扩散-呼吸的模型可推演出理论的沉积物呼吸氧同位素分馏系数为[5]:$ {\alpha }_{\text{SOR}}=\sqrt{{\alpha }_{\text{WCR}}}=\text{0.9894} $ ,与本实验的观测值$ {\alpha }_{\text{SOR}}= $ 0.9912接近. 然而,本实验观测到的沉积物分馏系数与基于美国普吉特海湾(Puget Sound)的沉积物暗培养实验观测值(-5‰ — -1‰)[15]有一定的差异. 当氧气在沉积物中被完全消耗时,并不会对上层水体的溶解氧同位素产生影响,即同位素分馏系数为0‰. 氧气全部被消耗的情况在沉积物有机质含量较高、呼吸作用速率较快或沉积物有机质表面结构复杂阻碍氧气向上层水体扩散等条件下更容易发生. 当氧气在沉积物中的有机质表面被完全消耗,且整个沉积物耗氧层中“完全耗氧”的比例较高,则会导致沉积物耗氧的表观分馏系数由大约 -10‰升高至接近0‰. 因此沉积物耗氧的表观分馏系数应该在理论值-10.6‰与0‰之间. 美国普吉特海湾的沉积物的有机质含量的质量百分比为1%[15],远高于本实验长江口海域沉积物中的有机质含量(质量百分比约0.3%). 因此,高有机质含量的呼吸暗培养实验观测到的沉积物耗氧分馏系数更接近氧气被全部消耗的极限值0‰,而本实验的观测值更接近扩散-呼吸模型的理论值,即沉积物耗氧仍会对上层水体产生同位素分馏效应.综上,本实验观测到的
$ {\alpha }_{\mathrm{W}\mathrm{C}\mathrm{R}} $ 值在大洋生物的呼吸作用分馏系数范围内(0.974—0.986)[12],$ {\alpha }_{\mathrm{S}\mathrm{O}\mathrm{R}} $ 值与理论值接近,再次验证了实验方法的可靠性. 同时,实验结果表明长江口底层沉积物耗氧对溶解氧的分馏效应远小于发生在水柱中的呼吸作用耗氧. 因此,可以利用二者迥异的氧同位素分馏效应,区分并量化分析缺氧事件中水柱呼吸和沉积物呼吸的占比. -
过去对大洋溶解氧同位素的研究表明,大洋当中的氧同位素与氧浓度之间的表观分馏系数往往大于0.980,有时甚至接近或高于纯水柱呼吸作用氧同位素分馏系数的最高值. 这是由于大洋中的溶解氧同时还受到物理混合及沉积物呼吸作用的影响. 对于长江口沿岸,水体的缺氧往往伴随着表层的水华作用. 大量的有机质由海洋表层沉降至底层,水柱和沉积物中的海洋生物通过呼吸作用快速地分解有机物消耗氧气,造成水体缺氧. 若以混合层溶解氧的δ18O或δO2/Ar为初始值,以底层溶解氧的δ18O或δO2/Ar为呼吸消耗后的值,即可计算出特定站位由表层至底层溶解氧同位素变化的表观分馏系数
$ {\alpha }_{\text{APP}} $ . 此表观分馏系数可被视作是水柱呼吸和沉积物呼吸共同作用的结果. 基于本实验得到的两个呼吸作用端元值,和同位素质量守恒的原理,即可量化分析水柱和沉积物呼吸在整个耗氧过程中的占比. 氧同位素的质量守恒公式为:$ {f}_{\text{WCR}} $ 和$ {f}_{\text{SOR}} $ 分别为水柱呼吸和沉积物呼吸在总耗氧量中的占比,且$ {f}_{\text{WCR}}+{f}_{\text{SOR}}=1 $ . 因此,根据公式(12),沉积物耗氧占比的计算方式为:以Zhou 等[22]汇报的F6站位(126.00°E,30.60°N)为例,其底层海水的δ18O值分别为6.834‰,由氧氩比计算的氧浓度为160 μmol∙L−1. 假设该底层海水的初始溶解氧来源为等密度层海水在海洋混合层与大气达到平衡时的氧气,其δ18O值和氧浓度分别为0.7‰和264 μmol∙L−1. 根据混合层和底层的δ18O与相应的溶解氧饱和度的对数[
$ \ln\left(\text{[}{\text{O}}_{\text{2}}\text{]}/{\text{[}{\text{O}}_{\text{2}}\text{]}}_{\text{0}}\right) $ ]之间的斜率值,计算出F6站位耗氧过程的表观耗氧分馏系数$ {\epsilon }_{\text{APP}}= $ −12.3‰,对应$ {\alpha }_{\text{APP}}= $ 0.9877. 根据公式(13)及$ {\alpha }_{\text{WCR}} $ = 0.9791和$ {\alpha }_{\text{SOR}} $ = 0.9912,计算得出水柱耗氧的占比约为71%,沉积物耗氧为29%. 说明水柱耗氧是长江口F6站位的主要耗氧方式. -
本研究详细描述了海洋溶解氧的提取、纯化和同位素测量的流程. 介绍了氧同位素及气体比值的具体校正方式,并用实验室制备的第二标样验证了方法的准确性和可靠性. 通过对海水和沉积物进行暗培养,本研究测量出长江口特定的水柱呼吸和沉积物呼吸的分馏系数别为-20.9‰和-8.8‰. 二者之间的差异可被用作衡量这两种耗氧过程的端元值,结合同位素质量守恒的原则,即可实现对长江口缺氧区耗氧过程的量化分析. 基于已发表的数据和氧同位素质量守恒,推算长江口缺氧区的耗氧过程为水柱呼吸主导. 溶解氧同位素在各耗氧过程中的独特性质,使其成为示踪海洋氧循环复杂过程的重要指标之一,具有较大的应用前景.
海洋溶解氧同位素的测量方法及其在长江口缺氧研究中的应用
Oxygen isotope analysis of marine dissolved oxygen and its application to the Changjiang Estuary hypoxia
-
摘要: 海洋溶解氧及其同位素组成可有效示踪海洋氧循环过程中复杂的生物、化学和物理过程. 海洋溶解氧的采集及提取过程需在严格的真空条件下进行,以避免大气中氧气带来的影响. 本文详细阐述了利用特制的真空样品瓶采集海水,并在实验室真空管线上提取及纯化溶解气体的过程. 纯化后的溶解气体在气体同位素质谱仪上测量,经过零点校正、质量干扰校正和空气标样校正后得到高精度的氧同位素和氧氩比数据. 基于以上方法,本实验对长江口混合层海水及底层沉积物进行了呼吸作用暗培养. 通过在不同的时间节点采集暗培养的海水,并测量其溶解氧同位素组成. 基于实验结果,计算出长江口特征的水柱呼吸氧同位素分馏系数为-20.9‰,沉积物呼吸的氧同位素分馏系数为-8.8‰,二者具有明显的差异,可被用作区分及量化分析长江口不同耗氧机制的端元值. 结合长江口原位采集的底层溶解氧的同位素组成,及本实验确定的水柱呼吸和沉积物呼吸的氧同位素分馏端元值,基于呼吸过程中氧同位素分馏的质量守恒,计算出长江口F6站位(126.00°E,30.60°N)水柱耗氧占比约为71%,沉积物耗氧的比例约为29%,说明发生在水柱中的生物呼吸作用为长江口F6站位的主要耗氧机制.Abstract: Marine dissolved oxygen (DO) and its isotopic composition are useful tracers for the biogeochemical and physical processes in marine oxygen cycling. DO sampling and extraction need to be performed under strict vacuum conditions, in order to avoid contaminations from the atmospheric O2. In this study, we describe the detail procedures for DO sampling using vacuum sample flasks, and its extraction and purification processes using a lab vacuum line. The purified DO sample is analyzed on a gas mass spectrometer, isotopically corrected, and finally reported versus air. Based on this method, we performed dark incubation experiments using mixed layer seawater and basal sediments collected from the Changjiang Estuary. During the course of incubation experiments, we collected and analyzed the DO isotopic compositions of incubated water. Then, we calculated the oxygen isotopic fractionation factors for water column reparation (WCR) and sediment oxygen respiration (SOR), with values of -20.9‰ and -8.8‰, respectively. These two distinct fractionation factor endmembers could be used for quantifying the oxygen consumption budget for the Changjiang Estuary hypoxia. Based on the mass balance of oxygen isotope fractionations during respiration, we estimate that WCR contributes to about 71% and SOR contributes to about 28% of the total oxygen consumption at station F6 (126.00°E, 30.60°N) off the Changjiang Estuary. Therefore, we conclude that WCR is the dominate oxygen consumption process at station F6 during the Changjiang Estuary hypoxia.
-
Key words:
- dissolved oxygen /
- oxygen isotope /
- isotope measurement /
- the Changjiang Estuary /
- hypoxia
-

-
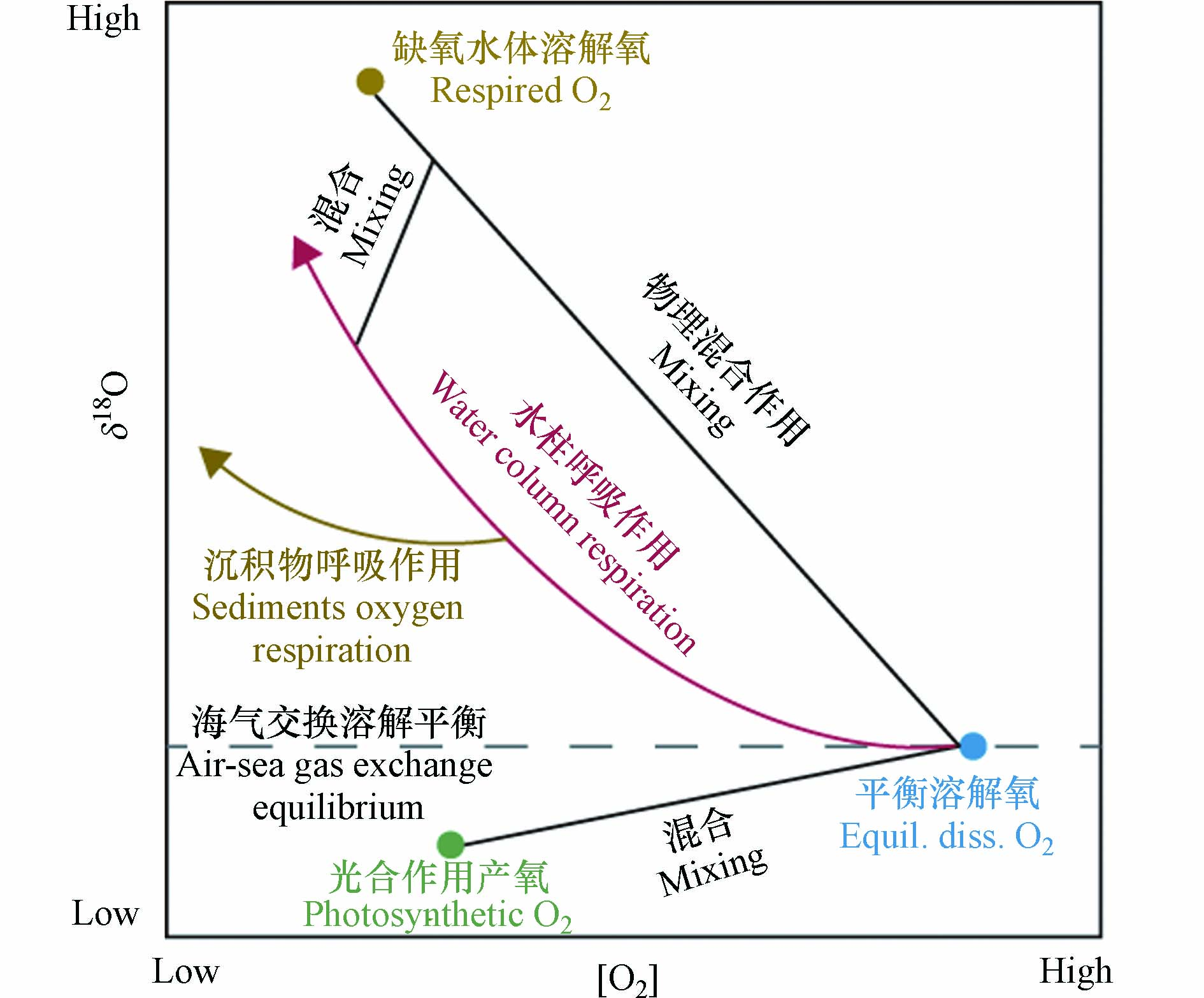
图 1 海洋氧循环过程中的同位素效应示意图
Figure 1. Schematic diagram of isotopic fractionations during marine oxygen cycling
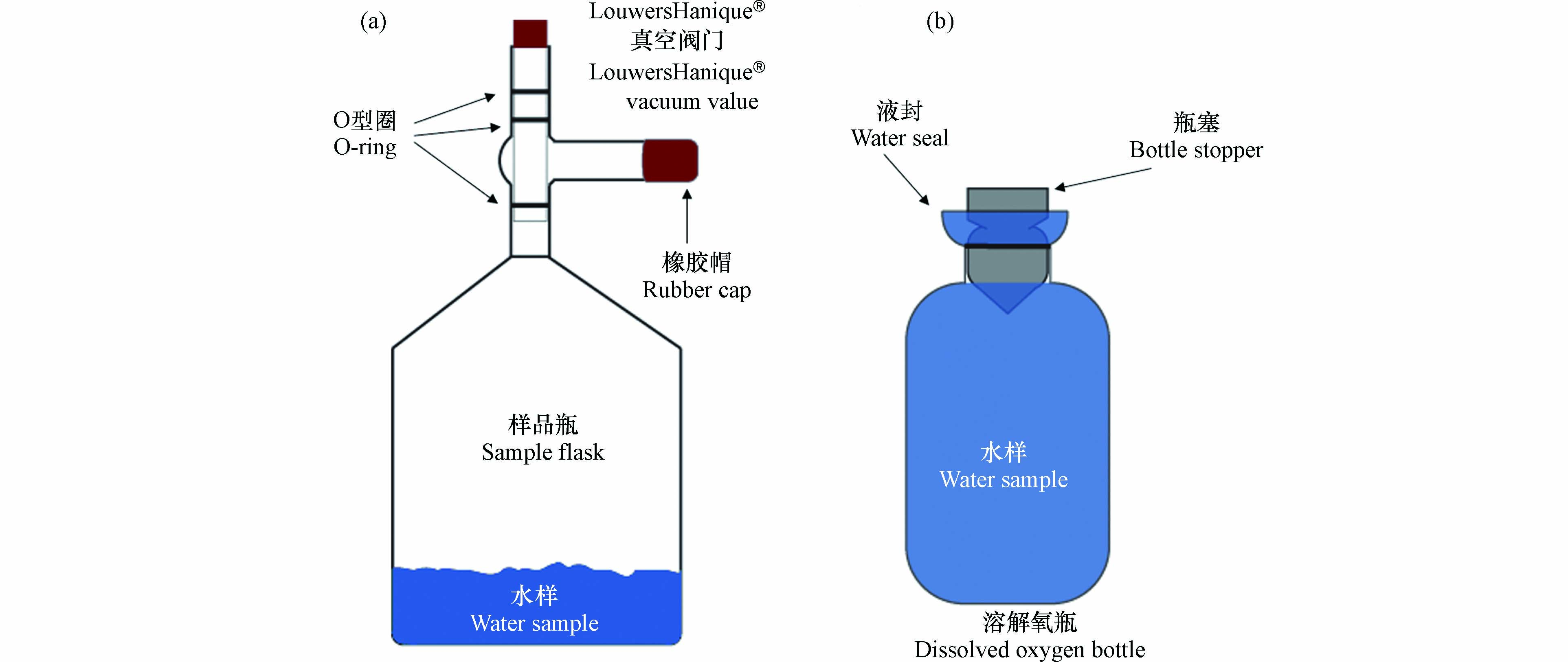
图 2 采集海洋溶解氧(a)的真空样品瓶和实验室呼吸作用暗培养所需的溶氧瓶(b).
Figure 2. Vacuum sample flask (a) for dissolved O2 collection, and dissolved oxygen bottle for dark incubation experiment (b).
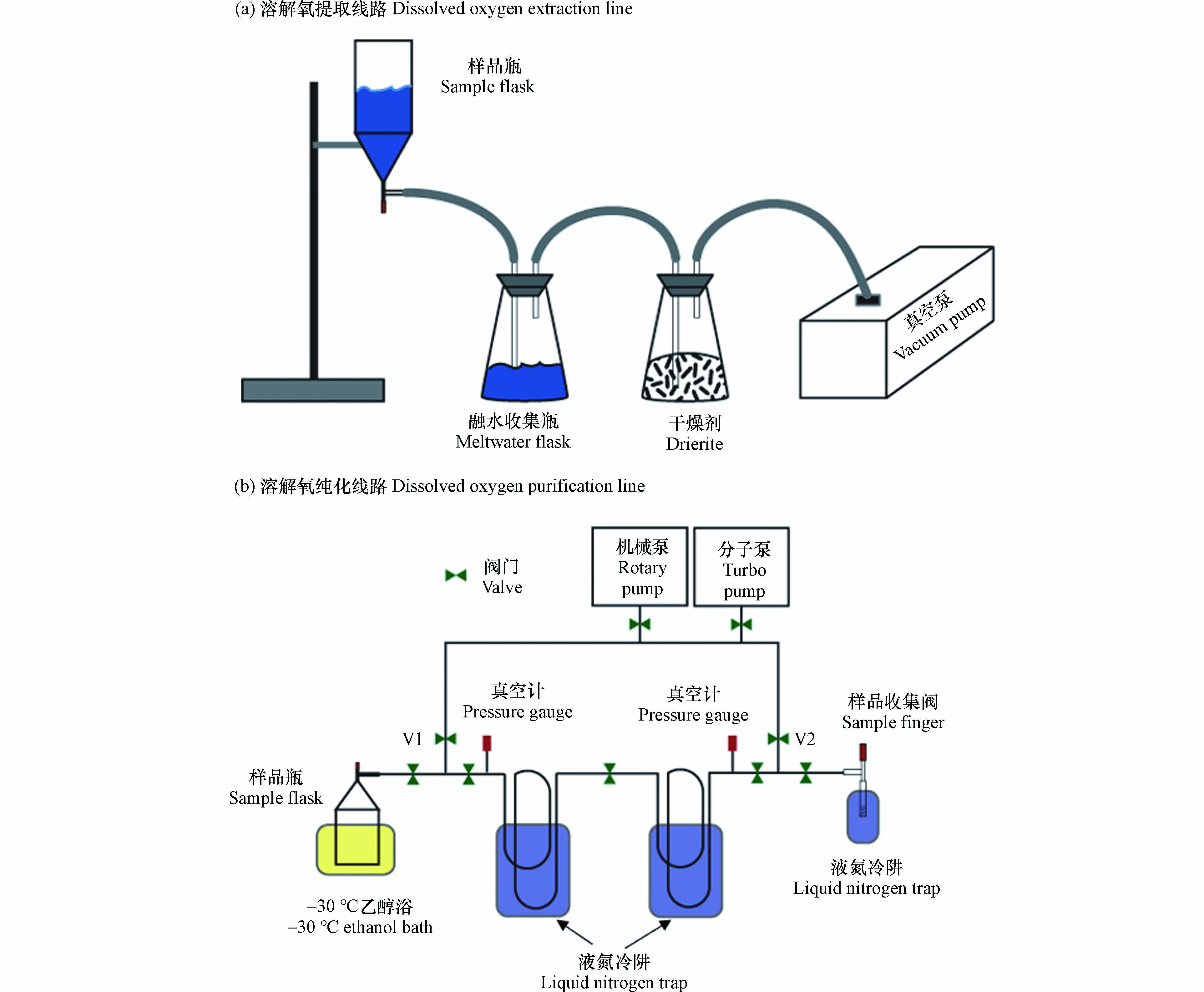
图 3 溶解氧的提取(a)和纯化(b)线路.
Figure 3. Dissolved oxygen extraction line (a) and purification line (b).
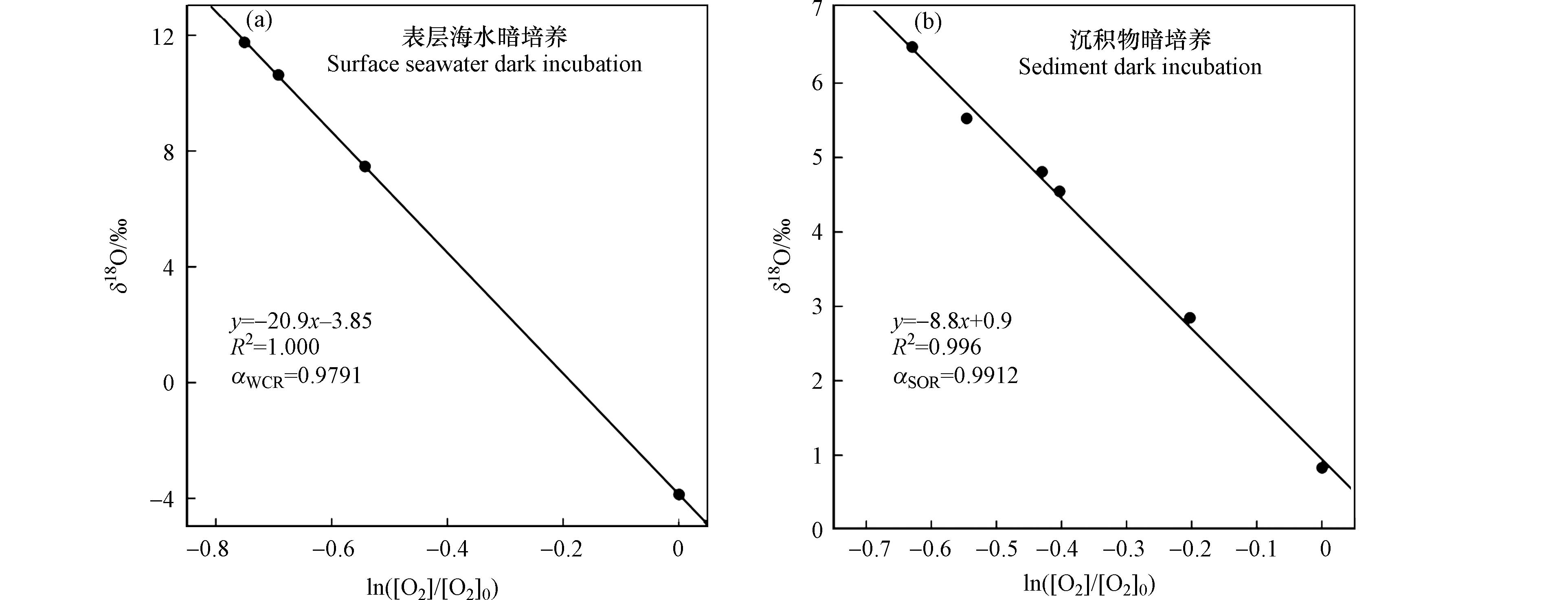
图 4 水柱呼吸和沉积物呼吸的氧同位素分馏系数
Figure 4. Oxygen isotope fractionation factor of water column respiration and sediment oxygen respiration
表 1 氧同位素测量校正参数及标样精度
Table 1. Isotopic correction parameters and precisions
2020.06 — 2020.10 2020.10—2021.03 零点校正a
Zero enrichmentNd 66 13 δ18O/‰ -0.005 -0.005 δO2/Ar/‰ +0.018 -0.011 质量干扰斜率
Mass interference slopeδ18O – δΝ2/O2 −5.514×10−5 −2.112×10−3 δO2/Ar – δΝ2/O2 −7.958×10−3 −1.341×10−2 空气标样精度b
Precision of air standardsNd 60 54 δ18O /‰ ±0.035 ±0.035 δO2/Ar /‰ ±0.4 ±0.5 平衡溶解气标样c
Dissolved gasesNd 7 5 δ18O /‰ 0.753 0.846 δO2/Ar /‰ -92.7 -90.8 a测量周期中所有零点校正的平均值;b空气标样精度为实验室测量的外部精度(1σ);c 实验室所制的平衡溶解气的氧同位素及气体比值平均值;d N代表标样数量
a Mean values of zero enrichments during the same analytical session; b External precisions (1σ) are established on air standards; c Mean values of lab equilibrated dissolved gases; d N represents numbers of analyses 下载: 导出CSV
下载: 导出CSV
表 2 培养实验取样时间及同位素数据
Table 2. Sampling information and isotopic data of dark incubation experiments
采样时间
Sampling timeδ18O / ‰ δO2/Ar / ‰ 溶解氧浓度/ (μmol∙kg−1)
Dissolved oxygen concentration混合层海水呼吸作用暗培养 2020.08.06a −3.858 29.9 259.4 2020.08.18 7.474 −401.4 150.8 2020.08.24 10.638 −484.7 129.8 2020.09.07 11.768 −514.0 122.4 沉积物呼吸作用暗培养 2020.09.15a 0.835 −125.9 210.1 2020.09.16 2.848 −293.0 169.9 2020.09.17 4.547 −406.3 142.7 2020.09.17 4.810 −427.3 137.7 2020.09.18 5.529 −490.9 122.4 2020.09.18 6.488 −530.8 112.8 a 该组培养实验的t0样品. a Initial incubation samples (t0)
下载: 导出CSV
-
[1] DIAZ R J, ROSENBERG R. Spreading dead zones and consequences for marine ecosystems [J]. Science, 2008, 321(5891): 926-929. doi: 10.1126/science.1156401 [2] STRAMMA L, SCHMIDTKO S, LEVIN L A, et al. Ocean oxygen minima expansions and their biological impacts [J]. Deep Sea Research Part I:Oceanographic Research Papers, 2010, 57(4): 587-595. doi: 10.1016/j.dsr.2010.01.005 [3] CHEN C C, GONG G C, SHIAH F K. Hypoxia in the East China Sea: One of the largest coastal low-oxygen areas in the world [J]. Marine Environmental Research, 2007, 64(4): 399-408. doi: 10.1016/j.marenvres.2007.01.007 [4] GONG H J, LI C, ZHOU Y T. Emerging global ocean deoxygenation across the 21st century [J]. Geophysical Research Letters, 2021, 48(23): 095370. [5] BENDER M L. The δ18O of dissolved O2 in seawater: A unique tracer of circulation and respiration in the deep sea [J]. Journal of Geophysical Research, 1990, 95(C12): 22243. doi: 10.1029/JC095iC12p22243 [6] LEVINE N M, BENDER M L, DONEY S C. Theδ18O of dissolved O2 as a tracer of mixing and respiration in the mesopelagic ocean [J]. Global Biogeochemical Cycles, 2009, 23(1): GB1006. [7] QUAY P D, EMERSON S, WILBUR D O, et al. The δ18O of dissolved O2 in the surface waters of the subarctic Pacific: A tracer of biological productivity [J]. Journal of Geophysical Research:Oceans, 1993, 98(C5): 8447-8458. doi: 10.1029/92JC03017 [8] MADER M, SCHMIDT C, van GELDERN R, et al. Dissolved oxygen in water and its stable isotope effects: A review [J]. Chemical Geology, 2017, 473: 10-21. doi: 10.1016/j.chemgeo.2017.10.003 [9] FRY B, BOYD B. Oxygen concentration and isotope studies of productivity and respiration on the Louisiana continental shelf, July 2007 [J]. Earth, Life and Isotopes, 2010: 223-241. [10] XI S S, LIU G J, ZHOU C C, et al. Assessment of the sources of nitrate in the Chaohu Lake, China, using a nitrogen and oxygen isotopic approach [J]. Environmental Earth Sciences, 2015, 74(2): 1647-1655. doi: 10.1007/s12665-015-4170-8 [11] ZHOU J, ZHU Z Y, HU H T, et al. Clarifying water column respiration and sedimentary oxygen respiration under oxygen depletion off the Changjiang Estuary and adjacent East China Sea [J]. Frontiers in Marine Science, 2021, 7: 623581. doi: 10.3389/fmars.2020.623581 [12] KIDDON J, BENDER M L, ORCHARDO J, et al. Isotopic fractionation of oxygen by respiring marine organisms [J]. Global Biogeochemical Cycles, 1993, 7(3): 679-694. doi: 10.1029/93GB01444 [13] SONG G D, LIU S M, ZHANG J, et al. Response of benthic nitrogen cycling to estuarine hypoxia [J]. Limnology and Oceanography, 2021, 66(3): 652-666. doi: 10.1002/lno.11630 [14] STOLPER D A, FISCHER W W, BENDER M L. Effects of temperature and carbon source on the isotopic fractionations associated with O2 respiration for 17O/16O and 18O/16O ratios in E. coli [J]. Geochimica et Cosmochimica Acta, 2018, 240: 152-172. doi: 10.1016/j.gca.2018.07.039 [15] BRANDES J A, DEVOL A H. Isotopic fractionation of oxygen and nitrogen in coastal marine sediments [J]. Geochimica et Cosmochimica Acta, 1997, 61(9): 1793-1801. doi: 10.1016/S0016-7037(97)00041-0 [16] OSTROM N E, GANDHI H, KAMPHUIS B, et al. Oxygen metabolism and water mass mixing in the northern Gulf of Mexico hypoxic zone in 2010 [J]. Geochimica et Cosmochimica Acta, 2014, 140: 39-49. doi: 10.1016/j.gca.2014.05.020 [17] ZHU Z Y, WU H, LIU S M, et al. Hypoxia off the Changjiang (Yangtze River) estuary and in the adjacent East China Sea: Quantitative approaches to estimating the tidal impact and nutrient regeneration [J]. Marine Pollution Bulletin, 2017, 125(1/2): 103-114. [18] ZHANG J, ZHANG Z F, LIU S M, et al. Human impacts on the large world rivers: Would the Changjiang (Yangtze River) be an illustration? [J]. Global Biogeochemical Cycles, 1999, 13(4): 1099-1105. doi: 10.1029/1999GB900044 [19] LI D J, ZHANG J, HUANG D J, et al. Oxygen depletion off the Changjiang (Yangtze River) Estuary [J]. Science in China Series D:Earth Sciences, 2002, 45(12): 1137-1146. doi: 10.1360/02yd9110 [20] PITCHER G C, AGUIRRE-VELARDE A, BREITBURG D, et al. System controls of coastal and open ocean oxygen depletion [J]. Progress in Oceanography, 2021, 197: 102613. doi: 10.1016/j.pocean.2021.102613 [21] ZHU Z Y, ZHANG J, WU Y, et al. Hypoxia off the Changjiang (Yangtze River) Estuary: Oxygen depletion and organic matter decomposition [J]. Marine Chemistry, 2011, 125(1/2/3/4): 108-116. [22] ZHOU F, CHAI F, HUANG D J, et al. Investigation of hypoxia off the Changjiang Estuary using a coupled model of ROMS-CoSiNE [J]. Progress in Oceanography, 2017, 159: 237-254. doi: 10.1016/j.pocean.2017.10.008 [23] 李宏亮, 陈建芳, 卢勇, 等. 长江口水体溶解氧的季节变化及底层低氧成因分析 [J]. 海洋学研究, 2011, 29(3): 78-87. doi: 10.3969/j.issn.1001-909X.2011.03.010 LI H L, CHEN J F, LU Y, et al. Seasonal variation of DO and formation mechanism of bottom water hypoxia of Changjiang River Estuary [J]. Journal of Marine Sciences, 2011, 29(3): 78-87(in Chinese). doi: 10.3969/j.issn.1001-909X.2011.03.010
[24] 韦钦胜, 王保栋, 陈建芳, 等. 长江口外缺氧区生消过程和机制的再认知 [J]. 中国科学:地球科学, 2015, 45(2): 187-206. doi: 10.1360/zd-2015-45-2-187 WEI Q S, WANG B D, CHEN J F, et al. Recognition on the forming-vanishing process and underlying mechanisms of the hypoxia off the Yangtze River Estuary [J]. Scientia Sinica Terrae, 2015, 45(2): 187-206(in Chinese). doi: 10.1360/zd-2015-45-2-187
[25] 吕晓霞, 翟世奎, 牛丽凤. 长江口柱状沉积物中有机质C/N比的研究 [J]. 环境化学, 2005, 24(3): 255-259. doi: 10.3321/j.issn:0254-6108.2005.03.007 LÜ X X, ZHAI S K, NIU L F. Study on the C/N ratios of organic matters in the core sediments of the Yangtze River Estuary [J]. Environmental Chemistry, 2005, 24(3): 255-259(in Chinese). doi: 10.3321/j.issn:0254-6108.2005.03.007
[26] YAO P, ZHAO B, BIANCHI T S, et al. Remineralization of sedimentary organic carbon in mud deposits of the Changjiang Estuary and adjacent shelf: Implications for carbon preservation and authigenic mineral formation [J]. Continental Shelf Research, 2014, 91: 1-11. doi: 10.1016/j.csr.2014.08.010 [27] SONG G D, LIU S M, ZHU Z Y, et al. Sediment oxygen consumption and benthic organic carbon mineralization on the continental shelves of the East China Sea and the Yellow Sea [J]. Deep Sea Research Part II:Topical Studies in Oceanography, 2016, 124: 53-63. doi: 10.1016/j.dsr2.2015.04.012 [28] QUIÑONES-RIVERA Z J, WISSEL B, JUSTIĆ D. Development of productivity models for the northern gulf of Mexico based on oxygen concentrations and stable isotopes [J]. Estuaries and Coasts, 2009, 32(3): 436-446. doi: 10.1007/s12237-009-9144-1 [29] LEHMANN M F, BARNETT B, GÉLINAS Y, et al. Aerobic respiration and hypoxia in the Lower St. Lawrence Estuary: Stable isotope ratios of dissolved oxygen constrain oxygen sink partitioning [J]. Limnology and Oceanography, 2009, 54(6): 2157-2169. doi: 10.4319/lo.2009.54.6.2157 [30] QUIÑONES-RIVERA Z J, WISSEL B, JUSTIĆ D, et al. Partitioning oxygen sources and sinks in a stratified, eutrophic coastal ecosystem using stable oxygen isotopes [J]. Marine Ecology Progress Series, 2007, 342: 69-83. doi: 10.3354/meps342069 [31] DePOORTER G L, ROFER-DEPOORTER C K. Oxygen-18 isotope effects in the electronic spectrum of UO2F2. II. Temperature effects [J]. Inorganic and Nuclear Chemistry Letters, 1978, 14(10): 333-335. doi: 10.1016/0020-1650(78)80050-6 [32] KROOPNICK P, CRAIG H. Oxygen isotope fractionation in dissolved oxygen in the deep sea [J]. Earth and Planetary Science Letters, 1976, 32(2): 375-388. doi: 10.1016/0012-821X(76)90078-9 [33] RAYLEIGH L. L. Theoretical considerations respecting the separation of gases by diffusion and similar processes [J]. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, 1896, 42(259): 493-498. doi: 10.1080/14786449608620944 [34] EMERSON S, QUAY P D, STUMP C, et al. Chemical tracers of productivity and respiration in the subtropical Pacific Ocean [J]. Journal of Geophysical Research, 1995, 100(C8): 15873. doi: 10.1029/95JC01333 [35] BENDER M L, TANS P P, ELLIS J T, et al. A high precision isotope ratio mass spectrometry method for measuring the O2N2 [J]. Geochimica et Cosmochimica Acta, 1994, 58(21): 4751-4758. doi: 10.1016/0016-7037(94)90205-4 [36] CASSAR N, BARNETT B A, BENDER M L, et al. Continuous high-frequency dissolved O2/Ar measurements by equilibrator inlet mass spectrometry [J]. Analytical Chemistry, 2009, 81(5): 1855-1864. doi: 10.1021/ac802300u [37] JURANEK L W, QUAY P D. Basin-wide photosynthetic production rates in the subtropical and tropical Pacific Ocean determined from dissolved oxygen isotope ratio measurements [J]. Global Biogeochemical Cycles, 2010, 24(2): GB2006. [38] OYABU I, KAWAMURA K, UCHIDA T, et al. Fractionation of O2∕N2 and Ar∕N2 in the Antarctic ice sheet during bubble formation and bubble-clathrate hydrate transition from precise gas measurements of the Dome Fuji ice core [J]. The Cryosphere, 2021, 15(12): 5529-5555. doi: 10.5194/tc-15-5529-2021 [39] HAMME R C, EMERSON S R. The solubility of neon, nitrogen and argon in distilled water and seawater [J]. Deep Sea Research Part I:Oceanographic Research Papers, 2004, 51(11): 1517-1528. doi: 10.1016/j.dsr.2004.06.009 [40] BENSON B B, KRAUSE D. The concentration and isotopic fractionation of oxygen dissolved in freshwater and seawater in equilibrium with the atmosphere [J]. Deep Sea Research Part B. Oceanographic Literature Review, 1984, 31(12): 859. [41] LI B D, YEUNG L Y, HU H T, et al. Kinetic and equilibrium fractionation of O2 isotopologues during air-water gas transfer and implications for tracing oxygen cycling in the ocean [J]. Marine Chemistry, 2019, 210: 61-71. doi: 10.1016/j.marchem.2019.02.006 [42] CRAIG H, HAYWARD T. Oxygen supersaturation in the ocean: Biological versus physical contributions [J]. Science, 1987, 235(4785): 199-202. doi: 10.1126/science.235.4785.199 [43] SPITZER W S, JENKINS W J. Rates of vertical mixing, gas exchange and new production: Estimates from seasonal gas cycles in the upper ocean near Bermuda [J]. Journal of Marine Research, 1989, 47(1): 169-196. doi: 10.1357/002224089785076370 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 4049
- HTML全文浏览数: 4049
- PDF下载数: 152
- 施引文献: 0









































