

-
汞是环境中毒性最强的重金属元素之一[1]. 它具有持久性、长距离迁移性和生物富集性,在自然界中能转化成剧毒的甲基汞,并通过食物链高度富集和放大,不仅对生态环境会产生严重危害,还时刻威胁着人类健康[2-3]. 2017年8月,旨在全球范围内控制和减少汞排放的《关于汞的水俣公约》正式生效,标志着全球携手减少汞污染迈出了关键一步. 我国长期以来被认为是汞生产、使用和排放最大的国家,因而面临巨大的履约压力[4]. 控制汞污染和汞排放,不仅要有完善的制度法规,还需要有准确的汞检测分析技术[5]. 海水中总汞含量低、测定过程中容易引入外源污染物且高盐基体可导致光谱干扰[6],稳定和精确测定海水中痕量总汞一直是环境监测领域亟待解决的难点之一[7].
目前测定水中总汞的国家和行业标准方法主要有冷原子吸收法、分光光度法和(冷)原子荧光法. 冷原子吸收法具有重现性好、灵敏度高等优点,但由于原子吸收光谱的限制,该方法线性范围较窄,对于海水中痕量总汞的测定,检出限并不理想[8];分光光度法一般采取双硫腙分光光度法,该方法灵敏度低,检出限高、并且操作复杂,现已较少使用[9];原子荧光测定总汞,优点在于灵敏度高、线性范围宽,但敞开式的直接液体进样方式容易导致样品的沾污、管路吸附导致汞的记忆效应明显、而且无法直接测定海水中痕量总汞[10].
本研究建立的方法是基于冷原子荧光光谱法,前端增加吹扫捕集装置,通过全封闭式顶空进样方式直接进入气态样品,避免样品沾污,同时实现汞与高盐基体的分离;利用对汞无吸附的特氟龙材质管路,降低汞的记忆效应;通过两级金捕获器进行金汞齐反应,实现汞的富集,从而显著降低方法检出限,提升灵敏度. 本研究开展了条件优化实验,确定了氧化效率最高且操作简洁的消解体系、最佳的还原剂用量和还原时效等重要参数. 在上述工作的基础上,对优化后的方法进行验证,获得了方法的检出限、测定下限、精密度、准确度等,特别是利用不同海区的多种海水样品对该方法的适用性进行验证,并与现行国标仲裁方法进行比较,以期得到一种稳定、精确测定海水中痕量总汞的方法.
-
痕量总汞分析仪(布鲁克斯兰德Ⅲ型冷原子荧光光谱仪及吹扫系统,美国)或者其他具有同等功能的仪器设备,原子荧光光度计(吉天SA-10,中国),电热消解仪(MARS 240/50,美国)、水浴锅(予华仪器HH-ZK4,中国)、磁力搅拌器(上海棱标85-1A,中国).
汞单元素标准物质(GBW08617,中国计量科学研究院,浓度为1000 mg·L−1,不确定度1 mg·L−1),汞标准样品(BY400030,坛墨质检,浓度为16.6 μg·L−1,不确定度1.2 μg·L−1).
其他试剂如硫酸、盐酸羟胺、氯化亚锡、高锰酸钾、硝酸、盐酸、溴化钾、溴酸钾、重铬酸钾均为优级纯,过硫酸钾为高纯.
-
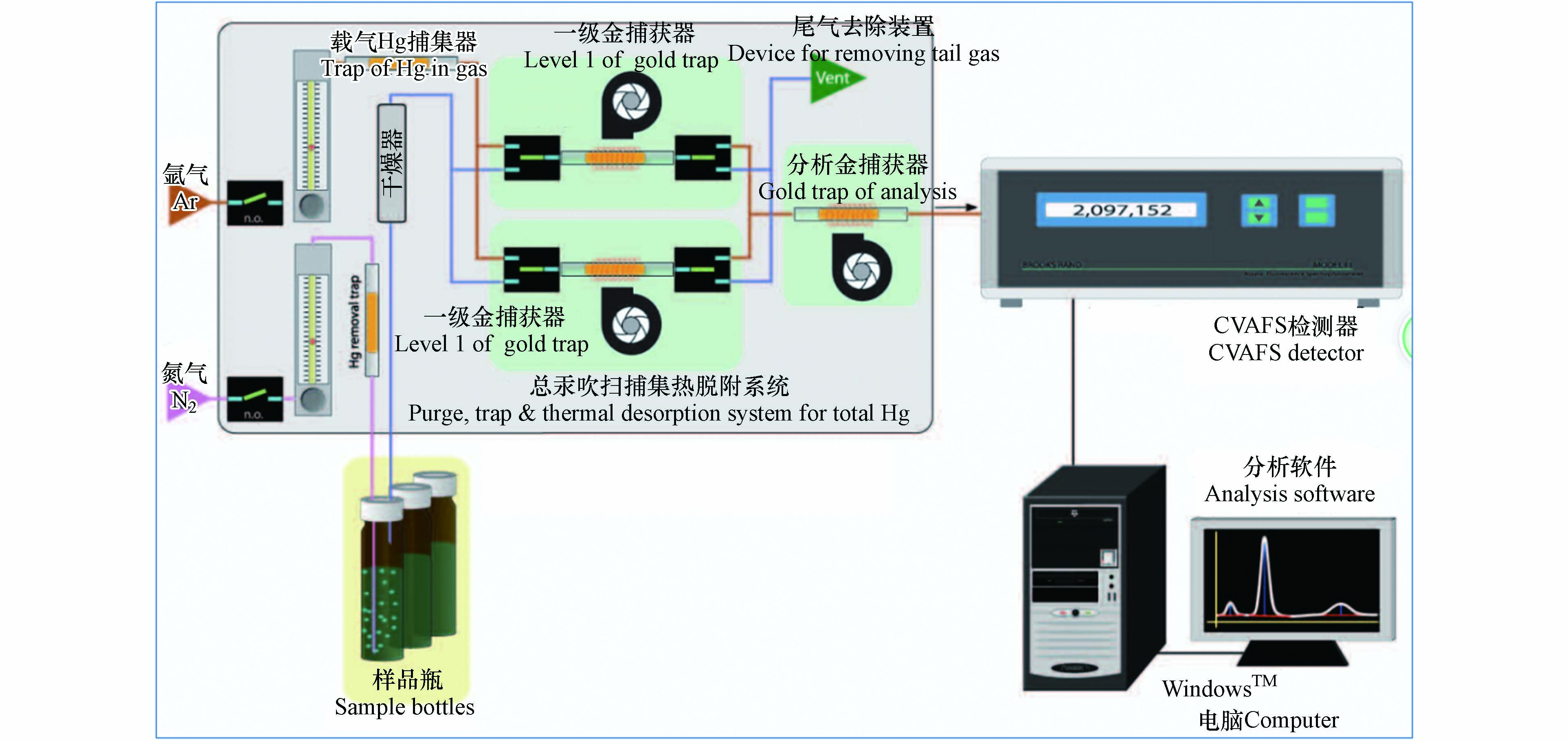
痕量总汞分析仪包括自动进样系统、吹扫捕集脱附系统和分析系统. 取15.0 mL制备好的样品放入(图1)具有聚四氟乙烯衬垫的玻璃材质样品分析瓶中,通过盐酸羟胺进行预还原,再通过氯化亚锡进行还原,立即盖紧瓶盖,进入分析. 利用氮气直接吹扫样品分析瓶中的样品,吹出的汞蒸汽首先通过干燥管干燥,之后被氩气带入一级金捕获器发生金汞齐反应,一级金捕获器快速升温,使汞迅速脱附,进入分析金捕获器进行金汞齐反应,分析金捕获器快速升温,汞再次脱附进入冷原子荧光光谱仪检测器产生信号值,仪器电脑软件对仪器信号值积分,峰面积与汞含量呈线性关系.
-
痕量总汞分析仪:PMT检测电压为540 V,载气(氩气)流量为45 mL·min−1, 吹扫气(氮气)流量为55 mL·min−1. 冷原子荧光的波长为253.7 nm, 峰面积积分. 出图时间为2.5 min,吹扫时间6.0 min,干燥时间3.0 min,热脱附时间为 2.5 min. 原子荧光分光度计:负高压:260 V,原子化高度:8 mm,灯电流:30 mA,载气流量(氩气):300 mL·min−1,屏蔽气流量:800 mL·min−1,读数时间:12 s,延迟时间:0.5 s,峰面积积分.
-
精确量取10.0 mL的1000 mg·L−1的汞单元素标准物质,用盐酸(1+199)溶液定容稀释至100.0 mL,制备成100.0 mg·L−1的汞标准储备液. 以盐酸(1+199)逐级稀释汞标准储备液,配制成1.00 μg·L−1的汞标准使用液. 取一定量的汞标准使用液于样品分析瓶中,定容至15 mL,配制成汞含量为0、50、100、500、1000 pg的标准系列溶液.
-
按照《海洋监测规范 第3部分:样品采集、贮存与运输》(GB 17378—2007)以及《近岸海域环境监测规范》(HJ 442—2020)的规定进行样品采集,采集好的水样加入硫酸保存,使样品pH<2,装于洁净的密封袋中. 此次实验的样品分别采自南海(1#样品)、东海(2#样品)、渤海(3#样品)、送检盲样(4#样品).
-
硫酸-高锰酸钾体系(SAPPS):取已硫酸酸化的15.0 mL水样加到50 mL具塞比色管中,依次加入0.37 mL的浓硝酸和0.60 mL的高锰酸钾溶液(50 g·L−1),保持水样成紫色,加塞摇匀,如15 min内紫色褪去,再补加高锰酸钾溶液(总用量不能超过4.5 mL),使水样维持紫色. 置于水浴中,于105 ℃消化1 h,取出冷却至室温. 临近测定时,边摇边滴加盐酸羟胺溶液(200 g·L−1),直至高锰酸钾刚好褪色为止,上机前加氯化亚锡溶液(200 g·L−1).
氯化溴体系(BCS):取2.5 L盐酸于烧杯中,中速搅拌,再加入 27 g 溴化钾溶解至少 1 h. 缓慢加入38 g 溴酸钾后,发生反应产生游离的卤素,溶液颜色黄变红变橘色,在通风柜中继续搅拌 1 h,使卤素散去,得到氯化溴试剂. 取已硫酸酸化后的水样15.0 mL,加入0.75 mL的氯化溴试剂,室温下放置10 min,样品颜色为黄色且滴加到淀粉碘化钾试纸上,淀粉碘化钾试纸变为紫色,否则应补加氯化溴试液(总用量不得超过2.4 mL),常温下消解24 h. 测试前滴加盐酸羟胺试液还原过剩的溴,使样品全部褪为无色,上机前加入氯化亚锡溶液.
盐酸-硝酸电热消解仪体系(HANAEDS):量取已硫酸酸化的水样25.0 mL放置于聚四氟消解管中,依次加入2.5 mL的浓盐酸和2.5 mL的浓硝酸,加塞摇匀,室温静置60 min. 然后放置于电热消解仪中,于180 ℃消解2 h. 取出后放冷至室温,将消解液转移至100 mL容量瓶中,用重铬酸钾硫酸溶液(取0.2 g重铬酸钾溶于900 mL水,再加27.8 mL浓硫酸,用水稀释定容至1000 mL)定容,上机前加入氯化亚锡溶液.
过硫酸钾-硫酸体系(PPSAS):量取已硫酸酸化的水样25.0 mL放置于125 mL样品瓶中,再加入1.25 mL过硫酸钾溶液(50 g·L−1),加热煮沸1 min后,冷却至室温,混匀. 测试前滴加盐酸羟胺溶液,上机前加入氯化亚锡溶液.
-
测定海水中总汞的原理是利用不同消解体系,把样品中不同价态的活性汞全部转化为相对稳定的二价汞,用盐酸羟胺还原过剩的氧化剂,再利用氯化亚锡将二价汞还原为单质汞. 不同消解体系对样品中不同价态的汞转化效率不同,故选择最优的消解体系是测定总汞的关键环节[11]. 参考国内外多种测定总汞的分析方法[12-14],综合比较后选择4种消解体系进行实验. 采用天然海水加标,加标浓度分为低、中、高组,分别为10、30、50 ng·L−1,探讨消解体系的氧化效率. 结果显示(图2),低浓度组,回收率从高到低依次为:过硫酸钾-硫酸体系>硫酸-高锰酸钾体系>盐酸-硝酸电热消解仪体系>氯化溴体系;中浓度组,回收率从高到低依次为:盐酸-硝酸电热消解仪体系>过硫酸钾-硫酸体系>硫酸-高锰酸钾体系>氯化溴体系;高浓度组,回收率从高到低依次为:盐酸-硝酸电热消解仪体系>过硫酸钾-硫酸体系>硫酸-高锰酸钾体系>氯化溴体系.
总体来看,硫酸-高锰酸钾体系、氯化溴体系和过硫酸钾-硫酸体系对不同浓度的加标样品具有稳定的氧化效率,组间差异较小. 盐酸-硝酸电热消解仪体系对于低、中、高组不同浓度的加标样品,氧化效率差异明显,方法稳定性不足. 这可能与该体系极强的氧化能力有关,一方面极强的氧化能力可能造成消解过程中汞的损失;另一方面,在环境监测领域,一般用盐酸-硝酸体系,辅助水浴、微波或者电热板等加热手段对土壤、沉积物、污泥以及植物等基体比较复杂的固态物质进行消解,回收率可达到91.1%—111.6%之间[11,15-16],说明该体系对基体较复杂的固体样品有很强的分解作用. 由于海水样品汞的分析采取非过滤形式[17],海水中并不均匀的悬浮颗粒物在强消解体系作用下被分解,使得回收率可能出现较大波动. 这并非汞样品分析中所期待的效果,本实验的目标是尽量降低不均匀的颗粒物的影响,因此该消解体系并不适合.
对比其他3组消解体系发现,氯化溴体系整体氧化效率最低,回收率在65.3%—80.2%之间. 氯化溴从回收率结果来看,与其他消解体系比较,并没有显示出较大优势,加之在配制氯化溴的过程中,会产生大量有毒溴气体,因而不是最优选择. 硫酸-高锰酸钾体系在各个浓度范围下,回收率均低于过硫酸钾-硫酸体系. 此外,硫酸-高锰酸钾体系中的高锰酸钾属于易制毒且易制爆试剂,处理起来并不安全,故也不推荐为最佳消解方法. 综合考虑,过硫酸钾体系在低、中、高浓度组的回收率均较高,且过硫酸钾非易制毒、易制爆试剂,较容易获得,为消解海水样品的最优消解体系.
-
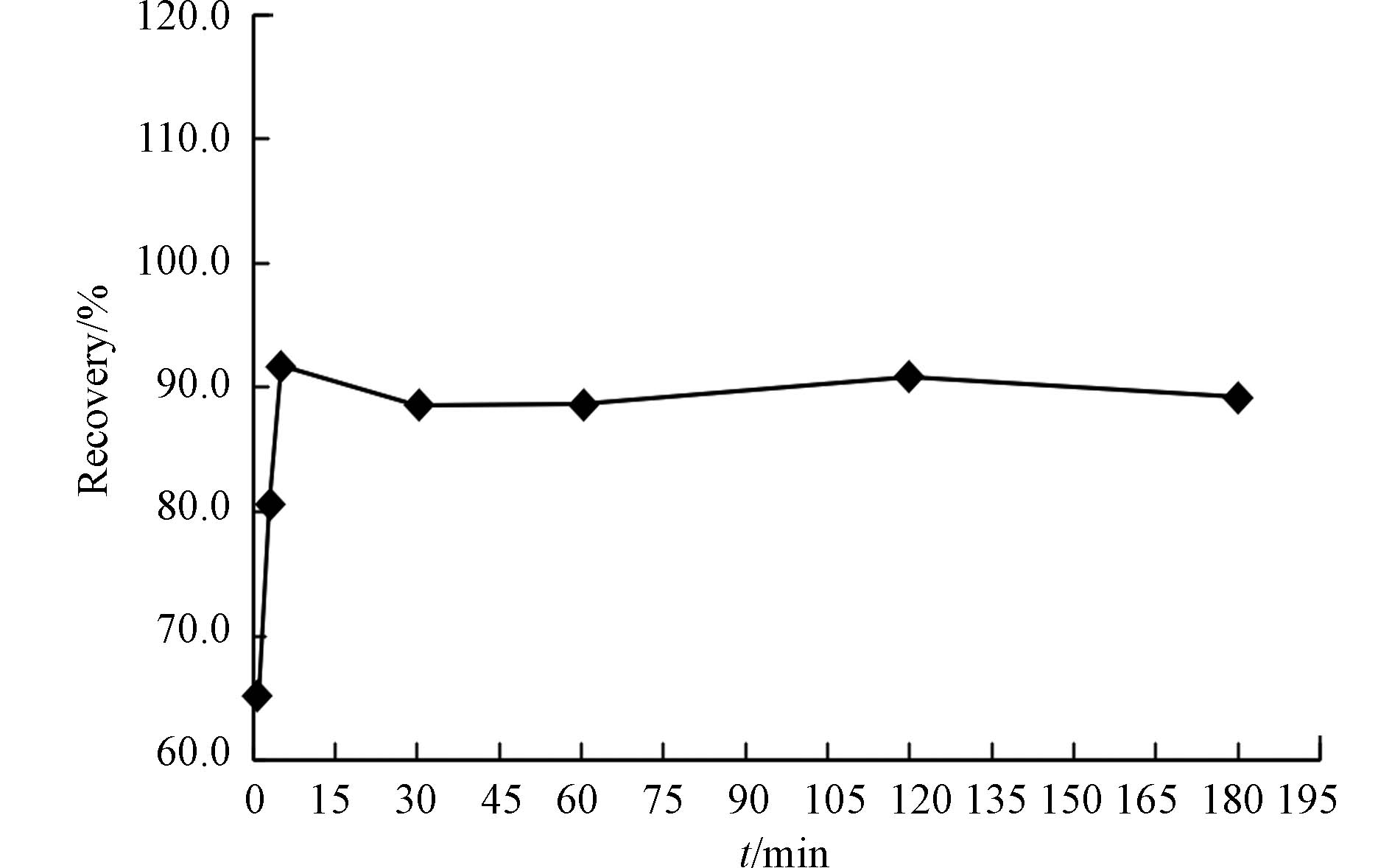
加入盐酸羟胺的主要作用是还原过量的氧化剂,例如在氯化溴体系中,盐酸羟胺的作用是还原氯化溴,去除卤素,为下一步汞离子还原为汞单质提供适当的还原性环境,还能防止金砂捕集管损坏;而在高锰酸钾-硫酸体系加入盐酸羟胺目的也是还原过量的高锰酸钾,否则过量的高锰酸钾同样会干扰汞的测定. 因此,盐酸羟胺的用量必须过量,不需要对盐酸羟胺的用量开展更多的研究. 但实验中常常无法确定加入盐酸羟胺后需放置多久才能把氧化剂除尽,故设计了1、3、5、30、60、120、180 min的7个时间点,用天然海水加标后的样品(加标浓度为30 ng·L−1)来考察盐酸羟胺的反应时间对回收率的影响情况(图3). 图3结果显示,加入盐酸羟胺5 min后,回收率即能达到91.8%,而后的各个时间点,其回收率并没有显著变化,只是在91.8%—88.6%范围内波动,且符合HJ 442对该量级的回收率要求,说明放置时间对回收率的影响并不大. 考虑到实验的用时和工作效率等问题,推荐加入盐酸羟胺后,应放置至少5 min方可进行下一步操作.
-
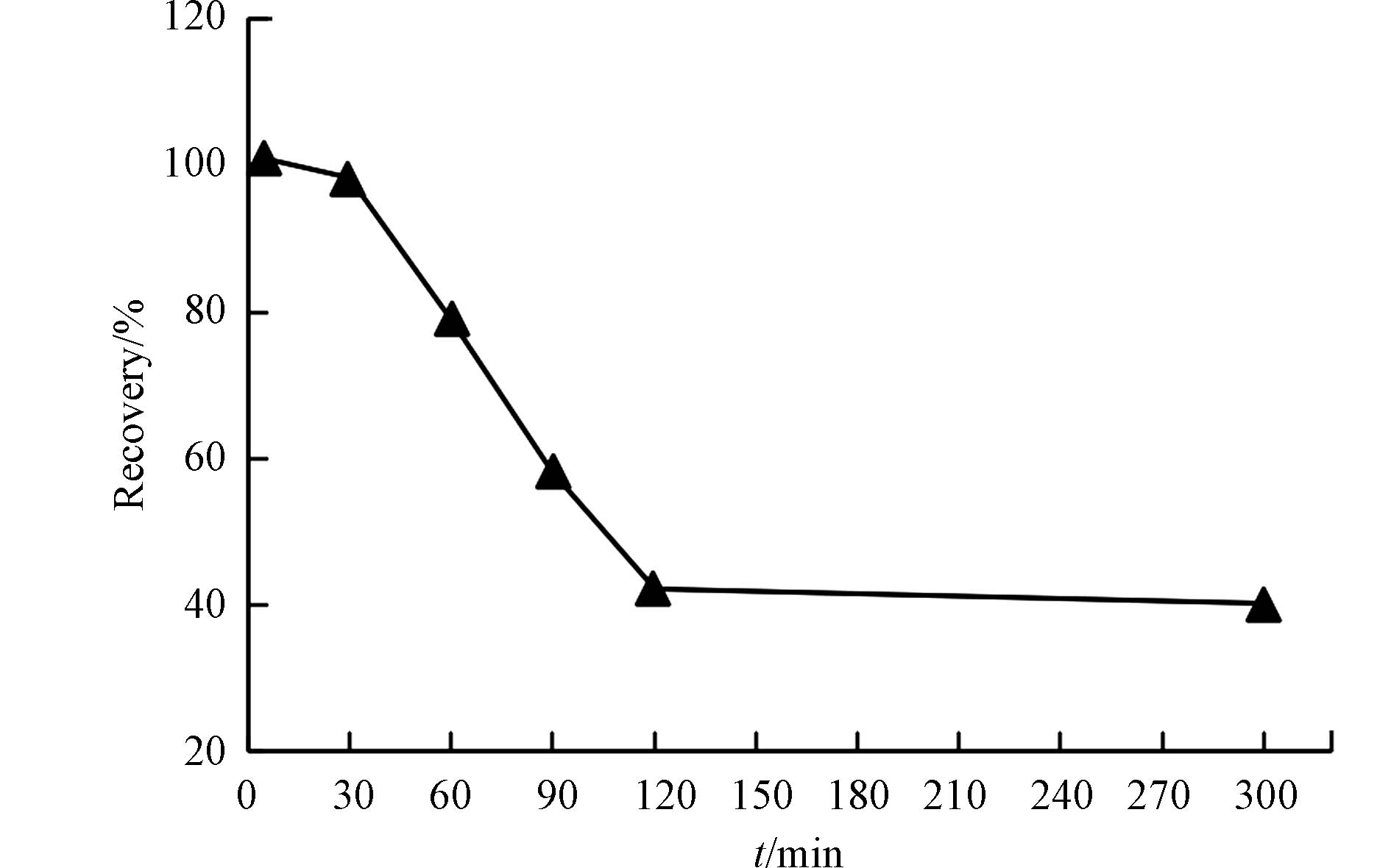
氯化亚锡是整个测汞环节中至关重要的一环,其作用是将样品中稳定的二价汞还原成具有挥发性的单质汞,由于单质汞具有挥发性,在氮气的吹扫下才会被顶空进样到金砂捕集管上进行汞的富集. 氯化亚锡的还原时间对其还原效率具有较大影响,然而还原时间的影响尚未有系统研究,通常都是即还原,即分析. 但是对于外部还原的样品,通常需要提前加入氯化亚锡,再通过仪器配备的自动进样器进样吹扫,此时控制还原时间以达到更高的还原效率就尤为重要. 本研究选取了6个时间点,分别是5、30、60、90、120、300 min,对同一天然海水加标样品(浓度为30 ng·L−1)进样分析,考察不同还原时间下,加标样品的回收率. 结果显示(图4),样品还原5 min,其回收率为101%,还原30 min,其回收率达到98.3%,但是超过30 min后,其回收率开始呈现显著下降的趋势,到60 min时,回收率仅为79.1%,到120 min时候,回收率接近40%,之后保持稳定. 综上考虑,样品加入氯化亚锡后务必在30 min内完成测定.
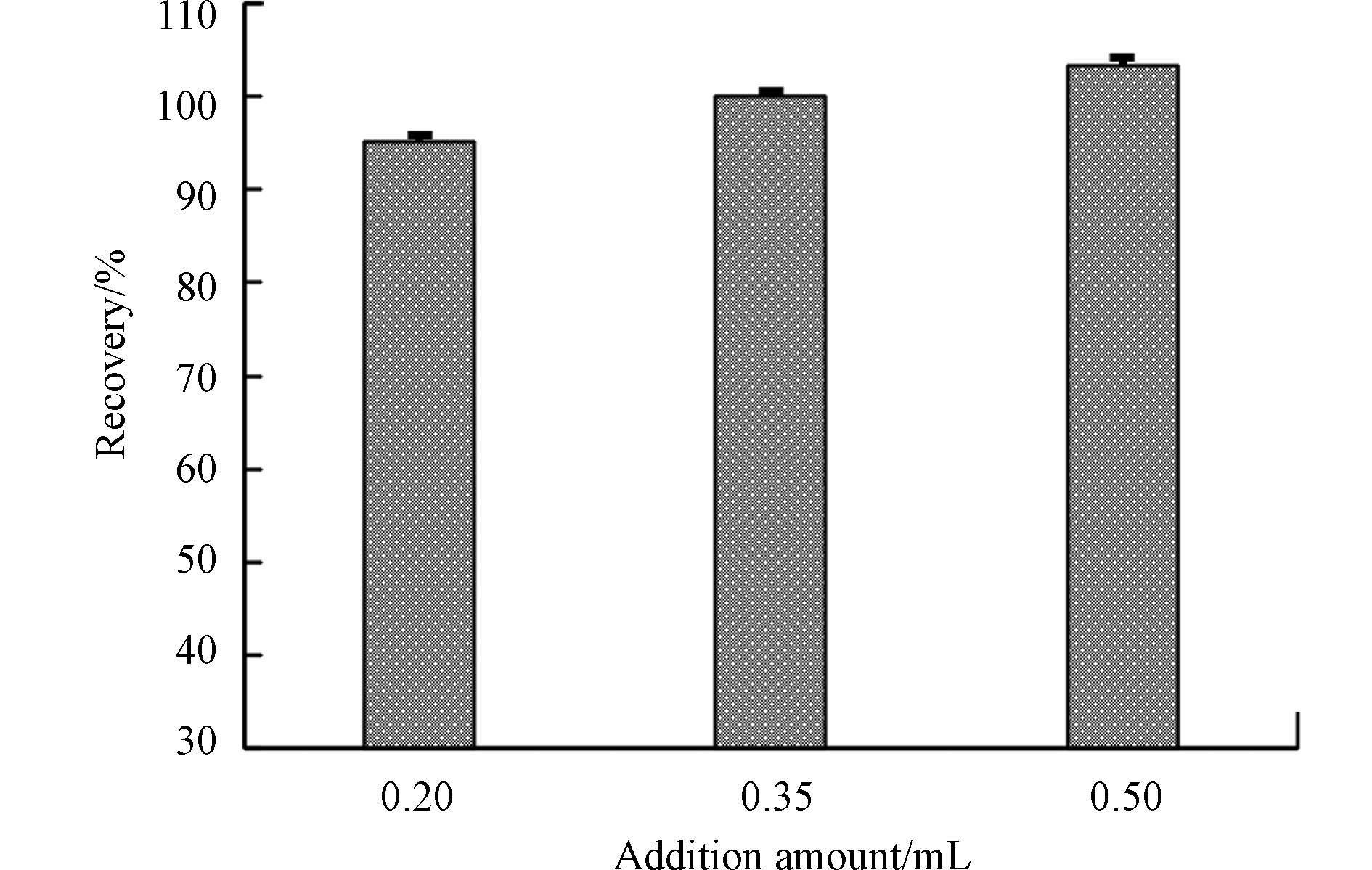
不同标准方法测定水中总汞时采用的氯化亚锡用量不同[13,18],为明确氯化亚锡用量对分析结果的影响,本文以浓度为55 ng·L−1的加标样品平行测定6次,来考察氯化亚锡用量对测定总汞的影响情况. 结果显示(图5),3种氯化亚锡用量条件下的回收率范围在95.2%—103%之间,未有显著性差异,说明氯化亚锡用量高于0.20 mL后对测定总汞的影响并不显著;表征精密度的相对标准偏差在1.3%—2.1%之间,0.35 mL氯化亚锡用量的相对标准偏差最小,因此将0.35 mL浓度为200 g·L−1的氯化亚锡作为还原剂用量的最佳条件.
-
选取实际海水样品,配置成加标浓度分别为15.0、35.0、55.0 ng·L−1的样品,进行方法准确度的验证. 上述浓度接近或者略高于我国近岸海域海水的实际状况,因此实验结果更有实用性. 各浓度均配制4个平行样,采用优化后的方法,按样品分析全步骤进行测定,并计算回收率. 结果显示(表1),汞的平均回收率范围在94%—104%,均符合HJ 442对该量级回收率的要求.
在15 mL的样品分析瓶中,分别加入汞标准使用液,使得样品瓶中的汞绝对量分别为100、500、1000 pg,对应的浓度为6.67、33.3、66.7 ng·L−1. 上述3种含量的样品,均按照优化后的方法,按样品分析全步骤,连续测定7次,验证方法的精密度. 结果显示(表2),低浓度样品(100 pg汞)相对标准偏差为3.6%,中浓度样品(500 pg汞)为2.6%,高浓度样品(1000 pg汞)为2.1%. 本方法对于低浓度样品的相对标准偏差控制在5%以内,完全符合HJ 442对精密度的要求.
-
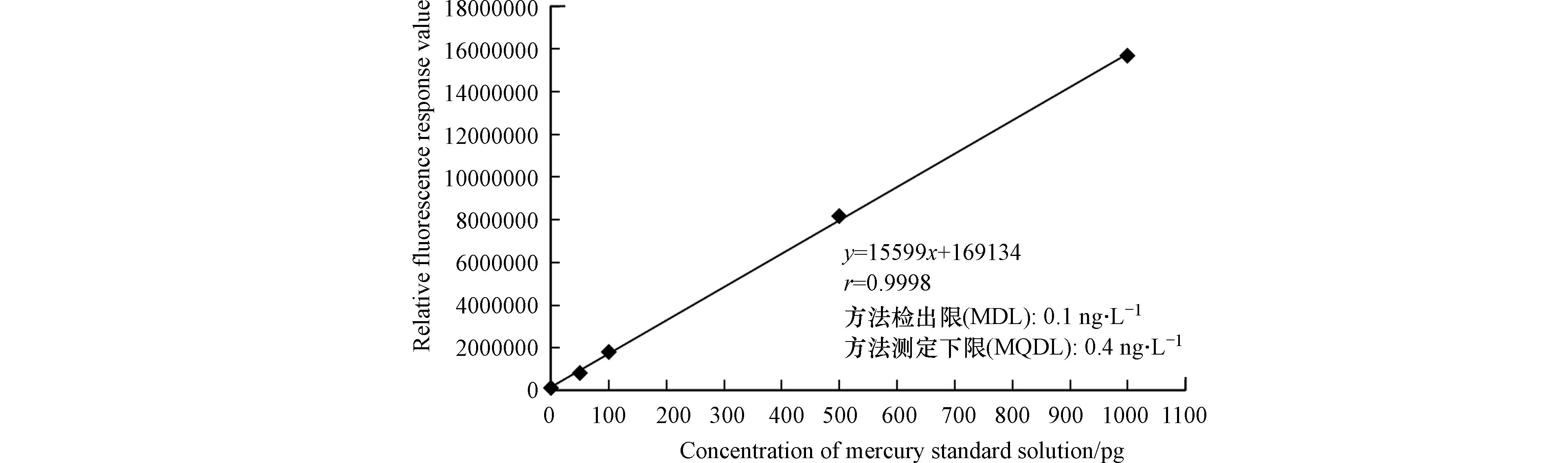
配置汞绝对量为0、50、100、500、1000 pg的标准系列,按照优化后的方法,依次进样,以汞绝对量为横坐标,荧光值强度为纵坐标,建立总汞测定的标准曲线,得到线性回归方程和r值,结果如图6所示. 结果显示,回归方程为y=15599x+169134,线性相关系数r大于0.995. 参照《环境监测分析方法标准制订技术导则(HJ 168—2020)》附录A方法,以实验用水做为空白,按优化后的方法,连续测定11次,计算标准偏差为0.035,进一步求算检出限为0.1 ng·L−1,以检出限的4倍作为测定下限,测定下限为0.4 ng·L−1.
进一步与目前已有的方法进行比较. 赵健等[19]建立了顶空固相微萃取-电热解塞曼原子吸收光谱法测定水中总汞,方法对水中总汞的检出限为30 ng·L−1;周巧丽等[20]利用活性炭定量吸附水中痕量汞,采用电热塞曼原子吸收光谱法测定活性炭富集的汞,检出限为0.3 ng·L−1;倪子月等[21]采用热解析富集装置对溶液中的汞富集后用X射线荧光光谱法进行检测,检出限为4 μg·L−1;高心岗等[22]利用氢化物发生-介质挡放电-原子荧光法测定汞的检出限为7.5 ng·L−1;赵小学等[23]建立了电感耦合等离子体-质谱法测定水中总汞,检出限范围在:1.15—31.1 ng·L−1;与上述已有的研究成果相比,本方法的检出限处于优先水平.
-
基于优化后的方法,对南海、东海、渤海以及送检未知的4类海水样品,进行汞含量分析,考察方法对实际样品的适用性. 每个样品平行分析6次,计算标准偏差和相对标准偏差. 结果显示(表3),实际样品分析结果的相对标准偏差范围为2.5%—10%,对于浓度仅为3 ng·L−1左右的样品,相对标准偏差也在10%,符合HJ 442对该浓度范围的相对标准偏差的要求.
-
采用本研究优化后的方法分析1#样品,并与2007版《海洋监测规范》(GB 17378—2007)中5.1章节原子荧光法测定海水汞的现行国标方法进行比较. 该方法是仲裁方法,其检出限为7 ng·L−1,测定下限为28 ng·L−1. 重复测定 6 次,结果显示,国标方法对于1#样品均未检出,而本研究方法测定1#样品的平均浓度为6.85 ng·L−1,RSD为0.51%(见表4). 1#样品低于国标方法的检出限,故1#样品用国标方法显示未检出,从表3中不难看出南海、东海、渤海以及送检未知的四类海水样品均低于国标方法的测定下限,说明本研究方法较现行国标方法在测定总汞含量在痕量级别的海水样品方面更具有优势. 取同一标准样品,分别用两种方法重复测定6次,得到国标方法的标准样品范围为(16.0—17.4)μg·L−1,RSD为3.6% ;本研究方法的范围为(16.4—17.2)μg·L−1,RSD为1.7%,两种方法均在标准值(16.6±1.2)μg·L−1范围内,符合要求,并无显著性差异.
-
本研究建立了一种基于吹扫捕集-冷原子荧光光谱法测定海水中痕量总汞的方法,并对水样的消解方法、盐酸羟胺的还原时间、氯化亚锡的还原时间和用量等前处理步骤进行了一系列优化. 过硫酸钾-硫酸消解体系的回收率不仅高而且稳定,消解过程安全且节约试剂,成为首选消解体系. 加入盐酸羟胺放置时间对结果影响较小,考虑到工作效率,建议盐酸羟胺的还原时间为5 min. 氯化亚锡的还原时间对结果有非常显著的影响,为了提升方法的效率,样品加入氯化亚锡后30 min内需要完成测定. 基于最优化的方法,对一系列参数进行验证,得到方法的线性为0.99995,检出限为0.1 ng·L−1、测定下限为0.4 ng·L−1 、回收率在94%—104%. 与国标方法(仲裁方法)相比较,国标方法测定实际海水样品为未检出,而本研究方法可以检出. 本研究方法具有检出限低、准确度高、多样品分析稳定性强等优势,能够用于海水中痕量总汞的分析.
基于氧化/吹扫捕集-冷原子荧光光谱法测定海水中痕量总汞
Determination of trace total mercury in seawater by oxidation/purge and trap-cold atomic fluorescence spectrometry
-
摘要: 基于氧化/吹扫捕集-冷原子荧光光谱法,建立了准确测定海水中痕量总汞的方法. 采用硫酸和过硫酸钾体系氧化海水样品,盐酸羟胺预还原,再经氯化亚锡将全部的汞离子还原为单质汞,原位吹扫和金汞齐捕集,在波长253.7 nm处原子荧光光谱法分析测定. 结果表明:该方法回收率为94%—104%,精密度相对标准偏差为2.1%—3.6%,方法检出限为0.1 ng·L−1,测定下限为0.4 ng·L−1,与现行国标方法相比,该方法增加了原位吹扫捕集的前处理步骤,因而灵敏度更高,检出限更低,也具有很好的稳定性. 用不同海区实际海水样品进行验证,结果显示,该方法满足实际样品的分析需求,建议在海水水质的汞监测领域能够拓展应用.
-
关键词:
- 吹扫捕集-冷原子荧光光谱法 /
- 海水 /
- 痕量总汞 /
- 痕量总汞分析仪.
Abstract: Based on the oxidation/purge and trap-cold atomic fluorescence spectrometry, a method for the accurate determination of trace total mercury in seawater was developed in this paper. The seawater sample was oxidized by sulfuric acid and potassium persulfate system, pre-reduced by hydroxylamine hydrochloride, and then reduced to elemental mercury by stannous chloride. The mercury ion was purged in the same position and trapped by gold amalgam. It was analyzed and determined by atomic fluorescence spectrometry at the wavelength of 253.7 nm. The results showed that the recovery was 94%—104%, the relative standard deviation of precision was 2.1%—3.6%, the detection limit was 0.1 ng·L−1, and the lower limit of determination was 0.4 ng·L−1. Compared with the current national standard method, the developed method has higher sensitivity, lower detection limit, and better stability. The method was validated by actual seawater samples from different sea areas, and the results showed that the method meets the analysis requirements of actual samples. Therefore, it is expected that the method can be widely used in the mercury determination of sea water. -

-
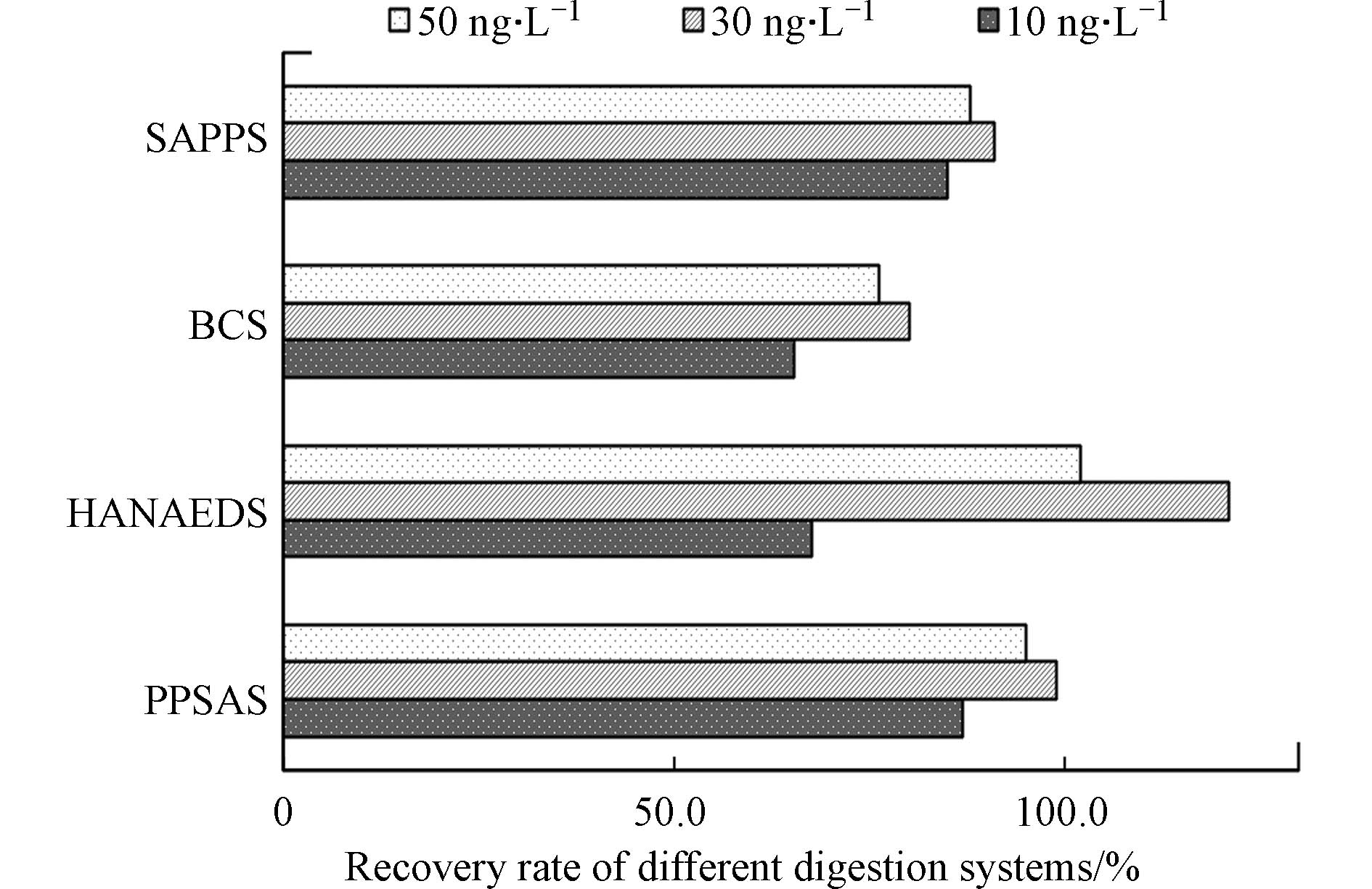
图 2 不同消解体系的回收率比较图
Figure 2. Comparison diagram of the recovery rates of different digestion systems
表 1 准确度结果
Table 1. Results of the accuracy
加标浓度/(ng·L−1)
Spiked concentration检测浓度/(ng·L−1)
Detection concentration平均值/(ng·L−1)
Average value回收率/%
Recovery1 2 3 4 15.0 20.6 20.4 21.2 21.3 20.9 104 35.0 41.1 39.7 42.8 39.3 40.7 101 55.0 54.0 57.2 55.9 60.4 56.9 94 注:本底浓度为5.30 ng·L−1  下载: 导出CSV
下载: 导出CSV
表 2 精密度结果
Table 2. Results of the precision
加入量/pg
Amount added测量值/pg
Detection value平均值/pg
Average value相对标准偏差/%
RSD1 2 3 4 5 6 7 100 93.2 98.4 102 101 104 97.5 104 99.9 3.6 500 485 477 491 488 498 519 501 494 2.6 1000 965 924 930 956 906 944 914 935 2.1
下载: 导出CSV
表 3 测定不同海区实际样品的结果
Table 3. Results of determination of actual samples from different sea areas
检测浓度/(ng·L−1)
Detection concentration平均值/(ng·L−1)
Average value标准偏差/(ng·L−1)
Standard deviation相对标准
偏差/%RSD1 2 3 4 5 6 1#样品 6.01 6.61 6.80 7.09 7.47 7.14 6.85 0.51 7.4 2#样品 3.88 3.85 3.95 3.16 3.57 3.10 3.59 0.38 10 3#样品 18.7 21.2 20.9 20.0 20.7 20.6 20.4 0.90 4.4 4#样品 10.6 10.5 10.7 10.1 10.9 10.5 10.6 0.27 2.5
下载: 导出CSV
表 4 实际样品与标准样品测定结果的对比
Table 4. Comparison of determination results between actual samples and standard samples
序号
Number实际样品测定/(ng·L−1)
Determination of actual samples标准样品测定/(μg·L−1)
Determination of standard samples本研究方法
The present method国标方法
The national standard method本研究方法
The present method国标方法
The national standard method1 6.01 ND 16.6 17.4 2 6.61 ND 16.7 16.9 3 6.80 ND 17.2 17.5 4 7.09 ND 16.7 16.0 5 7.47 ND 16.4 16.7 6 7.14 ND 16.4 16.4 平均值 6.85 — 16.7 16.8 相对标准偏差/% 0.51 — 1.7 3.6 注:ND为未检出;本研究方法为优化后的吹扫捕集-冷原子荧光光谱法;国标方法:《海洋监测规范》(GB17378.4—2007)中原子荧光法测定海水中汞的方法.
Note:ND represents No Detection; the present method is the optimized purge and trap-cold atomic fluorescence spectrometry; the national standard method is the atomic fluorescence spectrometry used to determine the mercury in sea water prescribed by “the Specification for Marine Monitoring” (GB17378.4-2007).
下载: 导出CSV
-
[1] 王晓晨, 李天鹏, 尤凯鸿, 等. 汞的多介质环境行为研究进展 [J]. 环境科学与技术, 2018, 41(12): 90-97. doi: 10.19672/j.cnki.1003-6504.2018.12.014 WANG X C, LI T P, YOU K H, et al. The progress in the research on multimedium environmental behaviors of mercury [J]. Environmental Science & Technology, 2018, 41(12): 90-97(in Chinese). doi: 10.19672/j.cnki.1003-6504.2018.12.014
[2] 陈亮, 李述贤, 杜譞, 等. 《关于汞的水俣公约》履约困境和出路 [J]. 中国生态文明, 2015(2): 74-79. CHEN L, LI S X, DU X, et al. Difficulties and solutions for“the Minamata Convention on Mercury” [J]. China Ecological Civilization, 2015(2): 74-79(in Chinese).
[3] 孟其义, 钱晓莉, 陈淼, 等. 稻田生态系统汞的生物地球化学研究进展 [J]. 生态学杂志, 2018, 37(5): 1556-1573. doi: 10.13292/j.1000-4890.201805.024 MENG Q Y, QIAN X L, CHEN M, et al. Biogeochemical cycle of mercury in rice paddy ecosystem: A critical review [J]. Chinese Journal of Ecology, 2018, 37(5): 1556-1573(in Chinese). doi: 10.13292/j.1000-4890.201805.024
[4] 郭卫广, 刘建国. 全球汞控制公约形势及中国履约需求分析 [J]. 环境污染与防治, 2010, 32(9): 107-111. doi: 10.3969/j.issn.1001-3865.2010.09.026 GUO W G, LIU J G. Perspective of global mercury control convention and obligation analysis for China [J]. Environmental Pollution & Control, 2010, 32(9): 107-111(in Chinese). doi: 10.3969/j.issn.1001-3865.2010.09.026
[5] 孙阳昭, 陈扬, 蓝虹, 等. 中国汞污染的来源、成因及控制技术路径分析 [J]. 环境化学, 2013, 32(6): 937-942. doi: 10.7524/j.issn.0254-6108.2013.06.003 SUN Y Z, CHEN Y, LAN H, et al. Study on pollution sources, cause of mercury pollution and its control technical roadmap in China [J]. Environmental Chemistry, 2013, 32(6): 937-942(in Chinese). doi: 10.7524/j.issn.0254-6108.2013.06.003
[6] 阎海鱼, 冯新斌, 商立海, 等. 天然水体中痕量汞的形态分析方法研究 [J]. 分析测试学报, 2003, 22(5): 10-13. doi: 10.3969/j.issn.1004-4957.2003.05.003 YAN H Y, FENG X B, SHANG L H, et al. Speciation analysis of ultra trace levels of mercury in natural waters [J]. Journal of Instrumental Analysis, 2003, 22(5): 10-13(in Chinese). doi: 10.3969/j.issn.1004-4957.2003.05.003
[7] 刘莹, 翟世奎, 张爱滨, 等. ICP-MS测定海水中溶解态痕量重金属-直接稀释法 [J]. 海洋学报, 2008, 30(5): 151-158. LIU Y, ZHAI S K, ZHANG A B, et al. Determination of dissolved heavy trace metals in seawater on ICP-MS-Direct dilution method [J]. Acta Oceanologica Sinica, 2008, 30(5): 151-158(in Chinese).
[8] 韦利杭. 冷原子荧光光度法测定海水中的超痕量汞 [J]. 分析化学, 1996, 24(2): 247. doi: 10.3321/j.issn:0253-3820.1996.02.003 WEI L H. Determination of ultra-trace mercury in seawater by cold atomic fluorescence spectrometry [J]. Chinese Journal of Analytical Chemistry, 1996, 24(2): 247(in Chinese). doi: 10.3321/j.issn:0253-3820.1996.02.003
[9] 蔡慧华, 彭速标. 痕量汞的测定方法进展 [J]. 理化检验-化学分册, 2008, 44(4): 385-390. CAI H H, PENG S B. Progress of methods for determination of trace amounts of mercury [J]. Physical Testing and Chemical Analysis (Part B:Chemical Analysis), 2008, 44(4): 385-390(in Chinese).
[10] 巢静波, 王茜, 王静如, 等. 吹扫捕集-原子荧光光谱与同位素稀释质谱法结合测定海水中痕量汞 [J]. 分析科学学报, 2021, 37(2): 165-170. doi: 10.13526/j.issn.1006-6144.2021.02.005 CHAO J B, WANG Q, WANG J R, et al. Determination of trace mercury in seawater by purge and trap-atomic fluorescence spectrometry combined with isotope dilution mass spectrometry [J]. Journal of Analytical Science, 2021, 37(2): 165-170(in Chinese). doi: 10.13526/j.issn.1006-6144.2021.02.005
[11] 李仲根, 冯新斌, 何天容, 等. 王水水浴消解-冷原子荧光法测定土壤和沉积物中的总汞 [J]. 矿物岩石地球化学通报, 2005, 24(2): 140-143. doi: 10.3969/j.issn.1007-2802.2005.02.009 LI Z G, FENG X B, HE T R, et al. Determination of total mercury in soil and sediment by aqua regia digestion in the water bath coupled with cold vapor atom fluorescence spectrometry [J]. Bulletin of Mineralogy Petrology and Geochemistry, 2005, 24(2): 140-143(in Chinese). doi: 10.3969/j.issn.1007-2802.2005.02.009
[12] 国家质量监督检验检疫总局, 中国国家标准化管理委员会. 海洋监测规范 第4部分: 海水分析: GB 17378.4—2007[S]. 北京: 中国标准出版社, 2008. General Administration of Quality Supervision, Inspection and Quarantine of the People's Republic of China, Standardization Administration of the People's Republic of China. The specification for marine monitoring-Part 4: Seawater analysis: GB 17378.4—2007[S]. Beijing: Standards Press of China, 2008(in Chinese).
[13] 中华人民共和国环境保护部. 水质 总汞的测定 冷原子吸收分光光度法: HJ 597—2011[S]. 北京: 中国环境科学出版社, 2011. Ministry of Environmental Protection of the People's Republic of China. Water quality-Determination of Total mercury-Cold atomic absorption spectrophotometry: HJ 5977-2011[S]. Beijing: China Environmental Science Press, 2011(in Chinese).
[14] EPA. Method 1631, Revision E: Mercury in water by oxidation, purge and trap, and cold vapor atomic fluorescence spectrometry[S]. US: 2002. [15] 罗国兵, 姜惟惟. 王水消解/冷原子吸收测定污泥总汞方法的改进 [J]. 中国给水排水, 2012, 28(6): 91-93. doi: 10.3969/j.issn.1000-4602.2012.06.024 LUO G B, JIANG W W. Improvement on method for determination of total mercury in municipal sludge with aqua regia digestion/CVAAS [J]. China Water & Wastewater, 2012, 28(6): 91-93(in Chinese). doi: 10.3969/j.issn.1000-4602.2012.06.024
[16] 王璐, 杨胜香, 李仲根, 等. 水浴消解-冷原子荧光法测定植物总汞的消解体系选择 [J]. 矿物岩石地球化学通报, 2022, 41(1): 151-155. doi: 10.19658/j.issn.1007-2802.2021.40.093 WANG L, YANG S X, LI Z G, et al. Selection A water bath-based digestion system combined with cold vapor atomic fluorescence spectrometry to determine total mercury in plants [J]. Bulletin of Mineralogy, Petrology and Geochemistry, 2022, 41(1): 151-155(in Chinese). doi: 10.19658/j.issn.1007-2802.2021.40.093
[17] 中华人民共和国生态环境部. 近岸海域环境监测技术规范 第三部分 近岸海域水质监测: HJ 442.3—2020[S]. 北京: 中国环境科学出版社, 2020. Ministry of Ecology and Environment of the People's Republic of China. Technical specification for offshore environmental monitoring Part 3 offshore seawater quality monitoring: HJ 442.3—2020[S]. Beijing: China Environmental Science Press, 2020(in Chinese).
[18] 中华人民共和国国家环境保护总局. 水质 汞的测定 冷原子荧光法: HJ/T 341—2007[S]. 北京: 中国环境科学出版社, 2007. State Environmental Protection Administration of the People's Republic of China. Water quality-Determination of mercury-cold atomic fluorescent spectrophotometry: HJ/T 341—2007[S]. Beijing: China Environmental Science Press, 2007(in Chinese).
[19] 赵健, 张林楠, 雷永乾, 等. 顶空固相微萃取-电热解塞曼原子吸收光谱法测定水中总汞 [J]. 分析化学, 2021, 49(8): 1393-1401. doi: 10.19756/j.issn.0253-3820.211133 ZHAO J, ZHANG L N, LEI Y Q, et al. Determination of trace total mercury in water by headspace solid phase microextraction and electropyrolytic zeeman atomic absorption spectrometry [J]. Chinese Journal of Analytical Chemistry, 2021, 49(8): 1393-1401(in Chinese). doi: 10.19756/j.issn.0253-3820.211133
[20] 周巧丽, 郭鹏然, 潘佳钏, 等. 活性炭富集-电热塞曼原子吸收光谱法测定水中痕量的汞 [J]. 分析化学, 2016, 44(8): 1270-1276. doi: 10.11895/j.issn.0253-3820.160247 ZHOU Q L, GUO P R, PAN J C, et al. Activated carbon enrichment combined with pyrolysis Zeeman atomic absorption spectroscopy for determination of trace amounts of mercury in water [J]. Chinese Journal of Analytical Chemistry, 2016, 44(8): 1270-1276(in Chinese). doi: 10.11895/j.issn.0253-3820.160247
[21] 倪子月, 程大伟, 刘明博, 等. 热解析富集-能量色散X射线荧光光谱法对溶液中汞的测定 [J]. 光谱学与光谱分析, 2022, 42(4): 1117-1121. doi: 10.3964/j.issn.1000-0593(2022)04-1117-05 NI Z Y, CHENG D W, LIU M B, et al. The detection of mercury in solutions after thermal desorption- enrichment by energy dispersive X-ray fluorescence [J]. Spectroscopy and Spectral Analysis, 2022, 42(4): 1117-1121(in Chinese). doi: 10.3964/j.issn.1000-0593(2022)04-1117-05
[22] 高心岗, 吴得福, 李洪杰, 等. HG-DBD-AFS测定地下水中铅砷汞 [J]. 环境工程, 2017, 35(s1): 192-194,50. GAO X G, WU D F, LI H J, et al. Determination of lead, arsenic and mercury in groundwater with HG-DBD-AFS [J]. Environmental Engineering, 2017, 35(s1): 192-194,50(in Chinese).
[23] 赵小学, 赵宗生, 王玲玲. 水中汞的电感耦合等离子体-质谱法测定 [J]. 中国测试, 2013, 39(6): 50-52. ZHAO X X, ZHAO Z S, WANG L L. Determining mercury in water by ICP-MS [J]. China Measurement & Test, 2013, 39(6): 50-52(in Chinese).
-
 点击查看大图
点击查看大图
计量
- 文章访问数: 2848
- HTML全文浏览数: 2848
- PDF下载数: 73
- 施引文献: 0
