

-
酸性废水中氨氮的处理处置一直以来都是水处理领域关注的重点[1-3],线路板加工、光伏发电和采矿等行业往往产生大量的酸性氨氮废水[2, 4],过量的含氮废水排放至水体中将对生态环境和人体健康造成不良影响,如水体富营养化,生成恶臭气味等. 尽管生物硝化/反硝化法[5]、化学沉淀法[6]、吸附法[7]、折点加氯法[8]以及它们的组合工艺已经运用于水体中氨氮降解[9],但以上方法都对pH值具有高度敏感性[9-10],不能直接运用于酸性氨氮废水处理. 因此,酸性氨氮废水的降解是一个亟待解决的水处理难题. 电化学高级氧化法因其pH适应性广、投加药剂少、二次污染少和绿色高效等特点受到广泛关注,并且得到一定的实际应用[11-13].
氨氮电化学氧化分为直接氧化和间接氧化[13-14],在酸性环境中氨氮以质子化的铵态氮为主,无法接近阳极表面进行直接氧化过程[1],以活性氯物种介导的间接氧化为主. 活性氯物种的生成能力取决于阳极析氯性能[13, 15],电化学原位利用水体中的氯离子生成两种类型的活性氯物种,分为游离氯(Cl2、HOCl或OCl−)和氯自由基物种(Cl·、ClO·等). 游离氯,作为活性氯物种中非自由基物种,可与氨氮反应生成氯胺类物质,并逐步转化为氮气或硝酸盐[16]. 活性氯物种中的自由基物种(Cl·和ClO·等)可迅速将氨氮氧化成·NH2,并经过一系列自由基链式反应,最终转化为氮气[17]. 然而,游离氯与氨氮反应速率受到pH影响,酸性条件下二者反应速率快速下降[1]. 近年来,大量研究表明氯自由基物种(如Cl·和ClO·)容易扩散至溶液中,对氨氮氧化展现出优越性能[1, 17-19]. 因此,如何实现酸性条件下游离氯物种向氯自由基物种定向转化,是酸性废水中氨氮快速降解的关键科学问题. 本课题组在考察数个电化学水处理工程现场后,发现电化学技术实际运用过程中存在以下问题:工程现场并未注重对阳极材料的选择,通常只通过成本或经验来进行简单选择,并未考察阳极材料对活性物种的生成情况及目标污染物的去除效能,也未考虑水质条件对阳极实际性能影响,水质条件(尤其是氯离子浓度)对电氧化体系的有效运行起着关键作用[13, 20]. 如何在含氯条件下,促进活性氯物种生成,强化氨氮去除,是如今很多工程现场亟待解决的技术问题. UV/氯研究表明[21],UV可使游离氯分解生成氯自由基物种. 因此,电氧化耦合UV体系有望通过活化体系中反应活性较低的游离氯,生成对氨氮氧化具有强反应活性的氯自由基物种,从而提高氨氮降解速率[1, 3].
本研究选取3种市场上常见的商业阳极:钌铱电极(材质为RuO2-IrO2镀层和Ti基底)、二氧化铅电极(材质为PbO2镀层和Ti基底)和锡锑电极(材质为SnO2-Sb镀层和Ti基底)构建电氧化体系(EO),并耦合UV构建电氧化耦合UV体系(EO/UV). 考察3种不同阳极电氧化及其耦合UV体系下氨氮降解差异,以及副产物氯胺[22]和硝酸盐[16]的生成情况. 并从以下两方面分析造成该差异的原因:(1)阳极材料的析氯性能,通过对阳极进行电化学测试,得出不同阳极在酸性环境中的极化曲线,从而判断阳极材料的析氯性能;(2)活性氯物种的生成情况,分析不同体系下活性氯物种的生成与转化,推测酸性氨氮废水处理中的UV协同强化机制;最后,通过处理金属冲洗废水和氨气吸收塔清洗废水两股实际酸性废水,验证阳极材料作为氨氮电氧化关键因素以及UV引入实现酸性氨氮废水快速降解;结合电极稳定性实验,估算3种阳极不同体系下处理1 kg NH4+-N阳极耗损费用,以期为高效处理酸性氨氮废水提供思路.
-
硫酸铵((NH4)2SO4),硫酸钠(Na2SO4),氯化钠(NaCl),次氯酸钠溶液(NaOCl,10%活性氯浓度),叔丁醇(Tert-Butanol,TBA),5,5-二甲基-1-吡咯啉-N-氧化物(5,5-Dimethyl-1-pyrroline N-oxide,DMPO)购自阿拉丁工业公司(中国上海). 所有试剂至少为分析级,使用时无需进一步净化,在整个研究过程中使用去离子水. 采用几何表面积为9 cm2(3 cm × 3 cm)的钌铱电极(Ti/RuO2-IrO2)、二氧化铅(Ti/PbO2)和锡锑电极(Ti/SnO2-Sb)(中国山西昌力特种金属有限公司)作为电化学反应器的阳极和不锈钢(Stainless Steel,SS)(中国河北腾峰金属材料有限公司)作为电化学反应器的阴极. 紫外光源(275 nm,20 W)购于珠海市天辉电子有限公司.
TOC-VCPH总有机碳分析仪,日本岛津公司;pH计,上海仪电科学仪器股份有限公司;GPD-3303S直流电源,台湾固纬电源有限公司;CHI 760E电化学工作站;Bruker EMX A300 10/12(德国).
-
NH4+-N的浓度测定采用纳氏试剂分光光度法[23],在紫外可见分光光度计420 nm处测定. 采用离子色谱法(Dionex,USA)测定NO3−-N、NO2−-N和Cl−的含量. 采用N,N-二乙基-苯二胺法(DPD)检测反应中产生的游离氯(包括Cl2、HClO和ClO−)浓度和总氯(包括游离氯与氯胺类物质),并通过总氯与游离氯的差值来确定氯胺的量. 用DMPO作为捕获剂,在Bruker EMX A300 10/12(德国)上进行电子顺磁共振(electron paramagnetic resonance,EPR)分析. 利用TOC测定仪测定样品中总有机碳浓度(total organic carbon,TOC).
利用CHI 760E电化学工作站构建三电极体系,其中工作电极为Ti/RuO2-IrO2电极、Ti/PbO2电极和Ti/SnO2-Sb电极,辅助电极为饱和甘汞电极,对电极为SS电极. 线性扫描伏安法(Linear Sweep Voltammetry,LSV)测试条件为扫描电位1.0 V至2.4 V(vs. SCE)、扫描速率10 mV·s−1、溶液pH = 2.0. 若无另外说明,文中所有电压均以相对于饱和甘汞参比电极电位表示.
-
单位氨氮降解所需的能耗(energy consumption,EC)计算公式如(I)所示:
其中:EC为单位能耗(kWh), U为反应时电压值(V),I为反应时电流值(A),t为反应时长(min),c为污染物浓度(mg·L−1),V为反应溶液体积(L).
-
电极实际寿命[24](actual lifetime,ALT)计算公式如公式(II)所示:
其中:ALT 电极实际寿命(h), Iac为加速寿命实验反应电流密度(A·cm−2),tac为加速寿命实验反应时长(min),I为实际反应电流密度(A·cm−2).
-
氨氮电氧化体系降解实验在圆柱形石英电解槽中进行,由Ti/RuO2-IrO2、Ti/PbO2和Ti/SnO2-Sb 3种不同阳极和不锈钢阴极构成3种电化学系统,电极尺寸为30 mm × 30 mm × 1 mm,阴阳两极相互平行放置,电极极板之间相距15 mm. 降解实验在恒电流模式下进行,由直流电源控制电流密度. 反应体积为100 mL,含50 mg·L−1 NH4+-N,电解质为100 mmol·L−1 Na2SO4和20 mmol·L−1 NaCl,用H2SO4和NaOH将反应溶液pH调节到1.5、2.0、2.5,磁力搅拌器转速控制为500 r·min−1,反应时间为3 h,每隔30 min时间间隔取样. 所有实验至少进行3次,结果以平均值表示. 氨氮电氧化耦合UV降解实验则将电解槽改为100 mL正方形的石英电解槽,光源使用UV275 nm,其余实验条件与电氧化体系氨氮降解实验一致.
-
电极加速寿命实验在100 mL圆柱形石英电解槽中进行,由Ti/RuO2-IrO2、Ti/PbO2和Ti/SnO2-Sb 3种不同阳极和铂网阴极构成3种电化学系统,电极尺寸为10 mm × 10 mm × 1 mm,阴阳两极相互平行放置,电极极板之间相距1 mm. 降解实验在恒电流模式下进行,由直流电源控制电流密度. 反应体积为100 mL,电流密度为1.2 A·cm−2,电解质为1 mol·L−1 NaClO4,用H2SO4和NaOH将反应溶液pH调节到2.0,磁力搅拌器转速控制为500 r·min−1,使用万用表对电压进行实时监测.
-
为考察酸性环境下电氧化体系氨氮氧化性能,将溶液初始pH值调节为1.5、2.0、2.5进行实验. 图1a为Ti/RuO2-IrO2阳极电氧化及其耦合UV体系氨氮降解效果,电氧化体系下反应3 h后氨氮剩余浓度分别为33.59、30.93、26.96 mg·L−1(图1b);Ti/PbO2阳极电氧化体系为19.08、25.95、29.49 mg·L−1;Ti/SnO2-Sb阳极则为8.53、12.6、14.35 mg·L−1(图1c). Ti/RuO2-IrO2阳极电氧化去除率分别为35.5%、40.3%和47.0%;Ti/PbO2阳极电氧化体系氨氮去除率分别为62.1%、48.2%和41.5%;Ti/SnO2-Sb阳极氨氮去除率则为83.1%、75.0%和71.5%. Ti/SnO2-Sb阳极氨氮降解性能优于其他两种阳极,在pH = 2条件下氨氮反应速率常数分别为0.3922、0.3205、0.2992 min−1(图1d),为Ti/RuO2-IrO2阳极的2.30 — 4.56倍及Ti/PbO2阳极的1.25 — 3.00倍. 在酸性环境中氨氮以带正电荷的铵根离子存在,难以接近阳极表面,可以排除氨氮直接氧化的可能[25]. 因此,在酸性环境下氨氮降解是由活性氯物种介导的间接氧化过程,Ti/SnO2-Sb阳极因其析氯性能较好,可生成更多的游离氯与氨氮进行反应,故使得氨氮降解性能高于其余两种阳极. 不同阳极受到溶液pH值的影响具有显著差异,活性电极Ti/RuO2-IrO2氨氮降解性能随着pH值的下降而降低,并且在实验过程中发现活性电极Ti/RuO2-IrO2在酸性条件下会有一定的溶出现象,电极表面镀层脱落导致电极性能下降[26];惰性电极Ti/PbO2和Ti/SnO2-Sb氨氮降解性能则是随着pH值的下降而升高,这是由于在酸性环境中阳极析氧副反应受到抑制[27],有利于析氯反应的有效进行,使得氨氮降解性能有所提高. 不同电极材料的适用场景具有一定的区别. 因此,在实际运用中应更加全面的考虑电氧化体系中阳极材料的选择[13]. 然而,pH < 3时电化学氨氮去除效率仍然较低,主要是由于游离氯在酸性环境下与氨氮反应速率非常慢 [1, 8],在酸性环境中,游离氯与NH4+-N最快反应速率仅为104 mol·L−1·s−1[28].
研究报道UV/氯体系可以生成一系列活性氯自由基物种[21],因此,构建电氧化耦合UV体系进行实验. 图1a体现Ti/RuO2-IrO2阳极电氧化耦合UV体系在pH = 1.5、2.0、2.5氨氮去除率较电氧化体系提升2.02 — 2.48倍,图1b和c分别体现Ti/PbO2阳极和Ti/SnO2-Sb阳极电氧化耦合UV体系在3 h内均可实现氨氮去除,Ti/SnO2-Sb阳极通过UV协同作用在反应90 min即可实现氨氮去除,且不受初始pH值影响. Ti/PbO2阳极电氧化耦合UV体系较电氧化体系去除率提升1.67 — 2.40倍,Ti/SnO2-Sb阳极电氧化耦合UV体系较电氧化体系去除率提升1.70 — 2.12倍. 通过UV协同作用,3种阳极氨氮降解速率均有大幅度提高,推测是由于UV光辐射导致游离氯光解成HO·和Cl·,并在溶液中发生链式反应生成一系列活性氯自由基物种[21],使得氨氮降解由游离氯主导转为氯自由基物种主导. 氯自由基物种对氨氮选择性较高,反应速率更快,进而使得氨氮高效降解[18].
-
氨氮电化学氧化过程中,控制副产物形成是一个挑战. 氨氮氧化过程除了生成理想的氮气外,还会生成有害的副产物,如氯胺、亚硝酸盐和硝酸盐等. 氯胺在活性氯物种介导的氨氮氧化过程中不可避免的作为中间产物生成[22],其在溶液中生成与转化高度依赖活性物种的种类与反应动力学[19]. 图2a对比了3种不同阳极材料电氧化及其耦合UV体系在pH = 2下氨氮氧化过程中氯胺的积累情况,电氧化体系下,Ti/RuO2-IrO2阳极、Ti/PbO2阳极和Ti/SnO2-Sb阳极氯胺浓度分别为25.5 mg·L−1(按Cl2计算,下同)、24.5 mg·L−1和42.2 mg·L−1. 其中,Ti/SnO2-Sb阳极氯胺浓度高于其余两种阳极,是由于在反应过程中生成更多的游离氯与氨氮反应,导致更多的氯胺积累. 值得一提的是,相同阳极电氧化耦合UV体系和电氧化体系中生成氯胺浓度呈现显著差异. Ti/RuO2-IrO2阳极电氧化体系通过UV协同作用后,氯胺浓度从25.5 mg·L−1下降到5.1 mg·L−1,Ti/PbO2阳极和Ti/SnO2-Sb阳极则分别从24.5 mg·L−1下降到6.2 mg·L−1和42.2 mg·L−1下降到10.8 mg·L−1. 这种差异可能是氨氮氧化过程中活性物种改变造成的,氯胺积累情况可以反应游离氯生成水平[22],氯胺作为游离氯与氨氮反应的中间产物,体系中生成游离氯浓度越高,氯胺积累量就越多. 在UV协同作用下,将游离氯光解为羟基自由基和氯自由基,使体系中游离氯浓度下降,减少游离氯与氨氮反应生成氯胺. 同时UV光解生成的氯自由基物种与氨氮具有高反应活性,可选择性与氨氮反应生成氮气,使得相同阳极电氧化耦合UV体系氯胺浓度相较于电氧化体系有所降低.
UV/氯胺研究表明[29],氯胺的直接光解过程会生成氯自由基物种,在实际水处理工程中,废水往往含有有机质,有机质与氯自由基物种反应可能会导致氯代副产物的生成,这些氯代副产物往往具有更高的生物毒性[30]. 但是,当废水中同时有氨氮存在的时候,氯胺光解生成的氯自由基物种与氨氮具有良好的反应选择性,在高效降解氨氮的同时能够抑制氯代副产物的生成[24]. 电氧化耦合UV体系相较电氧化体系可以更有效去除氨氮,并控制消毒副产物的生成,具有较好的工程应用前景.
所有反应体系反应结束后,均未检测到亚硝酸盐;硝酸盐生成的占比如图2b所示,Ti/RuO2-IrO2阳极电氧化及其耦合UV体系反应结束硝酸盐生成的占比为分别为11.8%和7.5%,Ti/PbO2阳极和Ti/SnO2-Sb阳极则分别为12.3%和7.0%、14.4%和9.1%. 通过UV协同作用,不同阳极对N2的选择性有一定的提高. 说明随着UV的引入,能在一定程度上控制副产物的生成.
-
电极材料选择将直接决定电化学处理过程中污染物的去除效率,因为电极材料特性决定了反应类型,从而决定所涉及的电化学和化学机制[13]. 电化学氧化可以分为直接氧化和间接氧化. 直接氧化需要将污染物吸附到电极表面,通过直接电子转移来实现污染物降解. 间接氧化则依赖于在电极上产生的活性物质(HO·、Cl·等),以介导污染物的转化,活性物质生成受电极材料影响[15]. 实际废水中氯离子浓度较低,阳极表面析氧反应和析氯反应存在竞争且电位差异较小,削弱了电极析氯活性,不利于氨氮降解. 通过测定阳极极化曲线以及实际析氯性能来分析不同阳极氨氮氧化速率差异.
线性扫描伏安法可以分析电极的极化曲线,图3a、b、c测定3种电极在1 mol·L−1 NaCl和1 mol·L−1 NaClO4溶液中的极化曲线,可以看出3种电极在pH = 2条件下析氯电位和析氧电位. Ti/RuO2-IrO2:析氯起始电位为1.45 V,析氧起始电位为1.50 V;Ti/PbO2:析氯起始电位为1.52 V,析氧起始电位为1.70 V;Ti/SnO2-Sb:析氯起始电位为1.55 V,析氧起始电位为1.76 V. Ti/RuO2-IrO2电极由于析氧和析氯起始电位比较接近,导致其析氯过程伴随着严重的析氧副反应,析氯选择性较差. Ti/PbO2电极和Ti/SnO2-Sb电极析氯起始电位远低于析氧电位,因此析氯选择性较高[31]. 同时,通过分析两者的电势差来判断析氯性能[32]. 3种阳极电势差如下,Ti/RuO2-IrO2阳极:0.19 V;Ti/PbO2阳极:0.20 V;Ti/SnO2-Sb阳极:0.32 V. 该结果表明,阳极析氯性能为Ti/SnO2-Sb > Ti/PbO 2 > Ti/RuO 2-IrO2,该结论与电氧化体系氨氮降解规律吻合.
游离氯对氨氮有氧化作用[8],因此测定3种电极在电氧化及其耦合UV体系下生成游离氯的积累量(pH = 2.0). 如图3d所示,3种电极游离氯浓度分布:反应初始阶段迅速上升,经过一定时间后到达一个相对稳定的浓度. Ti/RuO2-IrO2电氧化体系和电氧化耦合UV体系中游离氯累积量(按Cl2计算,下同)分别为68.0 mg·L−1和58.0 mg·L−1,Ti/PbO2和SnO2-Sb则分别为74.0 mg·L−1与51.0 mg·L−1和145.0 mg·L−1与107.0 mg·L−1. Ti/SnO2-Sb阳极析氯能力远高于Ti/RuO2-IrO2电极和Ti/PbO2电极,电氧化体系下游离氯的数值的差异与氨氮氧化速率常数的差异高度一致(图1d). 有文献指出,尽管溶液pH值较低,但电极表面原位生成大量的游离氯有利于将氨氮氧化降解[33]. 同一阳极电氧化耦合UV体系和电氧化体系相比游离氯积累量有一定的下降,表明UV能够光解游离氯,使得溶液中游离氯的积累量有所下降. 但氨氮降解性能的差异较大,可以推测在电氧化耦合UV体系下,游离氯物种对于氨氮氧化过程并非起主要作用,而UV可以光解活性较低的游离氯生成高反应活性的Cl·和HO·. 因此,初步猜测电氧化耦合UV体系下自由基物种是氨氮氧化过程中关键活性物种.
-
由于游离氯在酸性条件下氧化氨氮的能力有限[1, 33],推测游离氯的UV光解产生了更多的活性自由基物种,包括HO·和氯自由基物种(例如,Cl·和ClO·). 相关研究报道,氯自由基物种是酸性溶液中有效去除氨氮的潜在物种[1, 3, 17-19]. 不同阳极电氧化及其耦合UV体系氨氮降解性能差异证实了该推论,即在UV存在的情况下,氨氮降解反应速率显著增加. 为了鉴别和比较不同阳极系统原位生成的氯自由基物种,在不同阳极电氧化及其耦合UV体系下进行EPR表征以及掩蔽实验.
图4a、b、c描述了不同体系中DMPO的相关信号. 在电氧化体系中,3种不同阳极系统只有归属于DMPO-HO·模式的信号峰可见,表明存在由阳极生成的HO·,当在反应体系中加入氨氮后,DMPO-HO·信号没有明显变化.
相反,在电氧化耦合UV体系中存在DMPOX和DMPO-ClO·信号,DMPOX是由DMPO被Cl·氧化形成的氧化衍生物,DMPO-ClO·是由DMPO被ClO·氧化形成的氧化衍生物[1, 19],表明氯自由基物种在UV协同下的电氧化耦合UV体系中普遍存在. 当随后向体系中加入氨氮时,相应的DMPOX和DMPO-ClO·信号消失,而DMPO-HO·模式保留. 这与之前研究发现很好地一致,与HO·相比,氨氮更容易被Cl·和ClO·氧化[10]. 加入4 mmol·L−1 TBA进行掩蔽实验,以识别可能的自由基物种. 不同阳极电氧化及其耦合UV体系掩蔽结果如图4d、e、f所示,TBA能同时猝灭HO·和Cl·. 不同阳极电氧化体系中加入TBA后,氨氮的去除率并未受到明显抑制,而不同阳极电氧化耦合UV体系中加入TBA后,对氨氮去除有显著影响. Ti/RuO2-IrO2阳极、Ti/PbO2阳极和Ti/SnO2-Sb阳极氨氮去除率分别下降了75.8%、69.2%和76.6%,表明氯自由基物种在电氧化耦合UV体系氨氮氧化过程中占据主要贡献. 进一步说明在电氧化体系下,氨氮氧化过程主要由游离氯发挥主要作用,而在电氧化耦合UV体系下,氨氮氧化过程主要由氯自由基物种介导.
-
通过EPR以及掩蔽实验证明在UV协同作用下生成了氯自由基物种(Cl·和ClO·). 文献报道Cl·和ClO·与NH4+-N具有极高的反应速率常数,分别是1.1 × 109 mol·L−1·s−1和3.1 × 109 mol·L−1·s−1,是HO·与NH4+-N反应速率常数的12.4倍和34.8倍[18],而HClO与NH4+-N的最快反应速率仅为105 mol·L−1·s−1[28]. 综上,同一电极电氧化及其耦合UV体系氨氮降解差异是由活性物种种类不同导致. 不同阳极氨氮降解性能差异则是由阳极析氯性能决定,游离氯物种作为氯自由基物种的前体物,其浓度也是氯自由基物种生成的关键因素[19]. 综合上述实验,电氧化体系下,电解H2O生成H+和OH−(反应式1),反应生成的OH−迁移到阳极表面电解生成表面羟基(MOx(HO·))(反应式2),Cl−被表面羟基(MOx(HO·))间接氧化生成Cl·(反应式3),Cl·可与Cl−反应生成Cl2•−(反应式4),Cl2•−与表面羟基(MOx(HO·))反应生成HClO(反应式5),两个Cl·结合反应生成Cl2(反应式6),Cl2水解生成HClO(反应式7). 阳极生成的HClO在电氧化体系氨氮氧化过程发挥主要作用,NH4+-N首先与HClO反应生成氯胺(反应式11-13),氯胺进一步被氧化生成N2或NO3−(反应式14-16). 电氧化耦合UV体系下UV将电氧化体系中原位生成的HClO光解为Cl·和HO·(反应式8),Cl·和HO·都可与HClO反应生成ClO·(反应式9-10),Cl·和ClO·作为活性氯自由基物种,可将NH4+-N反应选择性氧化为N2(反应式17-18).
-
为探讨不同体系处理实际酸性废水中氨氮的可行性,从深圳某印刷电路板制造产业园选取一种金属冲洗废水、一种氨气吸收塔清洗废水开展电化学处理实验. 具体水质条件如表1所示,金属冲洗废水中含有一定量的铜离子、盐酸、氨氮和TOC;氨气吸收塔清洗废水中含有一定量的硫酸、盐酸、氨氮及TOC. 为了避免铜离子降低溶液色度从而减少UV透射率,在进行实验前先进行电解沉积,去除水溶液中的铜离子.
图5a为金属冲洗废水中氨氮的去除情况,3种阳极电氧化体系对氨氮去除性能较差. 在3 h反应后Ti/RuO2-IrO2阳极和Ti/PbO2阳极氨氮浓度分别为33.09 mg·L−1和36.76 mg·L−1,仅完成34.44%和25.49%氨氮降解,Ti/SnO2-Sb阳极氨氮反应结束后氨氮浓度为16.88 mg·L−1,去除率为65.52%. 随着UV的引入,氨氮降解性能大幅提升,并在3 h内实现氨氮的完全去除. 图5b为氨气吸收塔清洗废水中氨氮的去除情况,3种不同阳极电氧化体系降解效果不佳. 在3 h反应后Ti/RuO2-IrO2阳极和Ti/PbO2阳极氨氮浓度分别为45.75 mg·L−1和48.42 mg·L−1,去除率仅为24.70%和18.74%,Ti/SnO2-Sb阳极则为23.27 mg·L−1,去除率则为59.92%. 而在电氧化耦合UV体系中,氨氮降解性能大幅提升,3种体系均可完成氨氮降解,较电氧化体系氨氮降解性能提高了1.52 — 5.33倍. 对比了3种商业阳极在酸性环境下处理废水中TOC的效率差异,两种废水TOC降解效果如下:金属冲洗水,Ti/RuO2-IrO2阳极电氧化及其耦合UV体系反应结束后TOC浓度为9.13 mg·L−1和10.27 mg·L−1,Ti/PbO2阳极为8.78 mg·L−1和9.57 mg·L−1,Ti/SnO2-Sb阳极则为7.92 mg·L−1和8.36 mg·L−1. 对于氨气吸收塔清洗水,Ti/RuO2-IrO2阳极电氧化及其耦合UV体系反应结束后TOC浓度为13.35 mg·L−1和14.59 mg·L−1,Ti/PbO2阳极为12.44 mg·L−1和13.35 mg·L−1,Ti/SnO2-Sb阳极则为10.98 mg·L−1和12.48 mg·L−1. 不同阳极间,TOC去除效率有一定的差异. Ti/SnO2-Sb阳极TOC去除率分别为Ti/RuO2-IrO2阳极和Ti/PbO2阳极的1.18 — 1.45倍和1.15 — 1.22倍. 这种差异可以通过阳极析氯性能来解释,Ti/SnO2-Sb阳极生成更多的游离氯与有机物反应,从而提高降解效率. 相同阳极电氧化体系及其耦合UV体系对两股实际废水中TOC处理效果相近,但电氧化体系较电氧化耦合UV体系TOC去除效率略高,推测是废水中的有机物与氨氮竞争导致. 3种阳极电氧化体系生成的游离氯对氨氮选择性不高,更多的游离氯与有机物反应;而电氧化耦合UV体系将游离氯物种转化为对氨氮选择性更高的氯自由基物种,能高效的对氨氮进行降解,水中的氨氮与有机物竞争,使得对有机物去除效率低于电氧化体系.
对不同体系中电化学能耗进行计算,电氧化及其耦合UV体系处理金属冲洗废水中1 kg NH4+-N 3种不同阳极电化学能耗如图5c所示,Ti/RuO2-IrO2阳极能耗分别为337.01 kWh和166.56 kWh,Ti/PbO2阳极能耗分别为567.30 kWh和94.46 kWh,Ti/SnO2-Sb阳极能耗分别为223.67 kWh和76.85 kWh;电氧化及其耦合UV体系处理氨气吸收塔清洗废水中1 kg NH4+-N 的3种不同阳极电化学能耗如图5d所示,Ti/RuO2-IrO2阳极能耗分别为605.34 kWh和183.40 kWh,Ti/PbO2阳极能耗分别为651.95 kWh和148.07 kWh,Ti/SnO2-Sb阳极能耗分别为269.96 kWh和130.51 kWh. 对于两种不同实际酸性废水处理,不同阳极电氧化耦合UV体系相较于电氧化体系能耗均有显著降低,说明电子利用率得到大幅提升.
-
选择合适的电极材料是实现高效、可持续、低成本电化学水处理的关键因素之一[15]. 单一去除效能并不能很好地反映电极在废水处理过程的实用性,应结合电极稳定性与成本进行评估. 用于本实验购入的3种商业阳极材料(1 m × 1 m × 2 mm)价格如下,Ti/RuO2-IrO2:5500元,Ti/PbO2:1500元,Ti/SnO2-Sb:1300元. 通过电极加速寿命实验(图6a),对电极稳定性进行评估,当电极电压高于9 V时,即认定电极失活[24]. 通过换算得出3种阳极在10 mA·cm−2条件下(pH = 2.0),电极寿命分别为Ti/RuO2-IrO2:53195 h(约6.07a),Ti/PbO2:128707 h(约14.69a),Ti/SnO2-Sb:229 h. Ti/PbO2在该条件下,电极寿命远高于其余两种电极材料. 虽然Ti/SnO2-Sb电极在氨氮废水处理上具有一定的优势,但其寿命远远低于其他两种电极. 因此,提高Ti/SnO2-Sb电极稳定性仍然是一个巨大的挑战[34].
将电极成本、寿命与氨氮降解性能相关联(图6b),可以估算氧化1 kg NH4+-N对应的阳极耗费,为实际工程阳极材料选择提供参考. 估算方式如下:通过氨氮氧化速率常数乘以阳极寿命得出阳极生命周期的氨氮降解量,再将氨氮降解量除以阳极成本,其数值被认为是氧化1 kg NH4+-N所需的阳极费用. 结果发现,由于UV协同作用下极大提高了氨氮降解速率,使得1 kg NH4+-N所需阳极费用大幅降低. 3种阳极材料中Ti/PbO2阳极成本最低,1 kg NH4+-N电氧化耦合UV体系为4.94 元,单独电氧化体系为10.38元,相较于Ti/RuO2-IrO2阳极降低10.24 — 13.05倍,Ti/SnO2-Sb阳极降低256.00 — 271.17倍. 综合来看,Ti/PbO2阳极是适用于酸性氨氮废水处理的优选电极材料. 当然,以上数据均在30 mm × 30 mm的阳极规模下获得的估算值,实际大型电化学反应器运行过程中,由于阳极面积、电化学工艺参数和水质条件的变化,电极综合性能会有所不同.
-
本文考察在酸性环境下3种商业阳极不同体系间氨氮降解效能差异,并通过对阳极极化曲线测试、实际游离氯积累量测定、EPR表征及掩蔽实验分析该差异. 结果表明:
1)电氧化体系下游离氯的生成对氨氮降解起着主要作用,不同阳极氨氮降解性能差异是由阳极析氯性能决定. 在光电体系下,氨氮降解主要是通过氯自由基物种介导,而游离氯物种作为氯自由基物种的前提物,其与电极析氯性能密切相关. 因此阳极析氯性能是酸性氨氮废水降解的关键因素之一.
2)电氧化耦合UV体系可实现酸性氨氮废水快速处理,相较于电氧化体系,3种不同阳极去除率提升了1.67 — 2.48倍、降解速率提升了1.55 — 2.78倍、副产物生成占比降低了1.57 — 1.75倍. 并且在两股实际酸性氨氮废水处理中,电氧化耦合UV体系相比电氧化体系展现了更优异的氨氮处理性能,氨氮去除率提升了1.52 — 5.33倍,电化学能耗降低2.06 — 6.00倍,实现实际酸性氨氮废水高效低能处理.
3)Ti/SnO2-Sb析氯性能远超其余两种阳极材料,但其电极寿命低. 结合电极寿命、成本与去除性能对比3种电极处理1 kg NH4+-N所消耗的费用,发现Ti/PbO2是酸性氨氮电化学处理的优选阳极材料,其具有良好的电极寿命,适用于长时间电化学系统的运行,为工程实际运用中阳极选择提供一定参考.
阳极材料对电氧化及其耦合UV处理酸性氨氮废水的影响分析
Effects of anode materials on ammonium removal performance using the electro-oxidation (EO) and the coupled UV/EO systems for acidic wastewater treatment
-
摘要: 线路板制造等行业生产加工过程中会产生酸性氨氮废水,传统的生物硝化/反硝化、物理分离、折点加氯等方法不适用于酸性条件处理. 电氧化体系具有pH适应性广、药剂投加量小、绿色高效等优势,是处理酸性氨氮废水的潜在应用技术. 电极材料是电化学处理氨氮废水的关键因素之一,系统探讨酸性水质影响下不同阳极的除氨性能具有实际指导意义. 本研究考察Ti/RuO2-IrO2、Ti/PbO2、Ti/SnO2-Sb 等3种商业电极在酸性环境下电氧化及其耦合UV体系降解氨氮性能差异,并通过分析造成差异的原因,推测酸性氨氮废水处理中UV协同强化机制. 结果表明:(1)阳极析氯性能是酸性氨氮废水降解的关键因素之一,Ti/SnO2-Sb阳极在pH = 2时游离氯积累量为145.0 mg·L−1,分别是Ti/RuO2-IrO2阳极与Ti/PbO2阳极的2.13倍及1.95倍,相对应其氨氮去除速率为Ti/RuO2-IrO2阳极的2.79倍,Ti/PbO2阳极的1.90倍;(2)UV的引入加速了酸性氨氮废水处理,电氧化耦合UV体系较电氧化体系氨氮降解速率提高了1.98 — 2.67倍,结合掩蔽实验和电子顺磁共振表征,证明氯自由基物种在电氧化耦合UV体系氨氮氧化过程起到关键作用;(3)3种电极电氧化耦合UV体系实现了金属冲洗废水和氨气吸收塔清洗废水两股实际酸性废水中氨氮的有效处理;(4)尽管Ti/SnO2-Sb阳极单次氧化氨氮性能最佳,但是其稳定性最差,通过比较3种阳极不同体系下去除1 kg NH4+-N的阳极耗损费用,发现Ti/PbO2相较其余两种阳极成本减少10.24 — 271.17倍.Abstract: Acidic wastewater laden with ammonia nitrogen (NH4+-N) is a widely produced in various industries, such as printed circuit board manufacturing. Due to the low pH level, traditional treatment methods like biological nitrification and denitrification, physical separation, and breakpoint chlorination are not practical for its treatment. Electrochemical methods have emerged as a promising approach for NH4+-N-containing wastewater treatment due to their wide range of pH adaptability, low reagent dosage, high efficiency, and eco-friendliness. Electrode material is a critical factor in determining the performance of electrochemical ammonia treatment, and it is essential to systematically investigate the effects of anode materials on NH4+-N removal performance under different acidic water conditions. In this study, we used three commercial electrodes (Ti/RuO2-IrO2, Ti/PbO2, and Ti/SnO2-Sb) in the electro-oxidation (EO) and coupled UV/EO systems and compared the performance of electrochemical ammonia removal at acidic pH values. The mechanism of UV-assisted NH4+-N removal in acidic wastewater was elucidated by investigating the differences between various electrodes and systems. The key findings can be summarized as follows. (1) The chlorine evolution capacity was identified as a critical factor influencing the EO of ammonia in acidic wastewater. Notably, the Ti/SnO2-Sb anode exhibited a significantly higher cumulative free chlorine of 145.0 mg·L−1 at pH = 2, which was 2.13 and 1.95 times greater than the corresponding values observed with the Ti/RuO2-IrO2 and Ti/PbO2 anodes, respectively. Consequently, the NH4+-N removal rate using the Ti/SnO2-Sb anode was 2.79 times and 1.90 times faster compared to the Ti/RuO2-IrO2 and Ti/PbO2 anodes, respectively. (2) The incorporation of UV irradiation accelerated the removal of NH4+-N in acidic wastewater. The coupled UV/EO system exhibited a 1.98 to 2.67 times higher NH4+-N removal rate compared to the EO system alone. Results from scavenging experiments and electron paramagnetic resonance tests demonstrated the significant role of chlorine radical species in NH4+-N removal within the coupled UV/EO system. (3) The coupled UV/EO systems using the three different anodes were found to be effective in removing NH4+-N in two actual acidic wastewaters generated from metal washing and ammonia-absorption-tower cleaning factories. (4) Although the Ti/SnO2-Sb anode exhibited the best performance in the single-stage oxidation of NH4+-N, it was the least stable among the three evaluated anode systems. Comparing the anode consumption cost for removing 1 kg of NH4+-N across different types of anodes, it was observed that Ti/PbO2 was significantly more cost-effective (with a cost reduction ranging from 10.24 to 271.17 times) compared to the other two anodes.
-
Key words:
- ammonia oxidation /
- anode materials /
- electrochemistry /
- UV coupling /
- active chlorine species.
-

-

图 1 不同pH影响下,3种阳极电氧化及其耦合UV体系中的氨氮降解效果(a)Ti/RuO2-IrO2;(b)Ti/PbO2;(c)Ti/SnO2-Sb以及(d)反应速率常数比较
Figure 1. Time courses of NH4+-N concentration in the EO and UV/EO systems with (a) Ti/RuO2-IrO2; (b) Ti/PbO2; and (c) Ti/SnO2-Sb anodes. (d) Comparisons of rate constants between different conditions
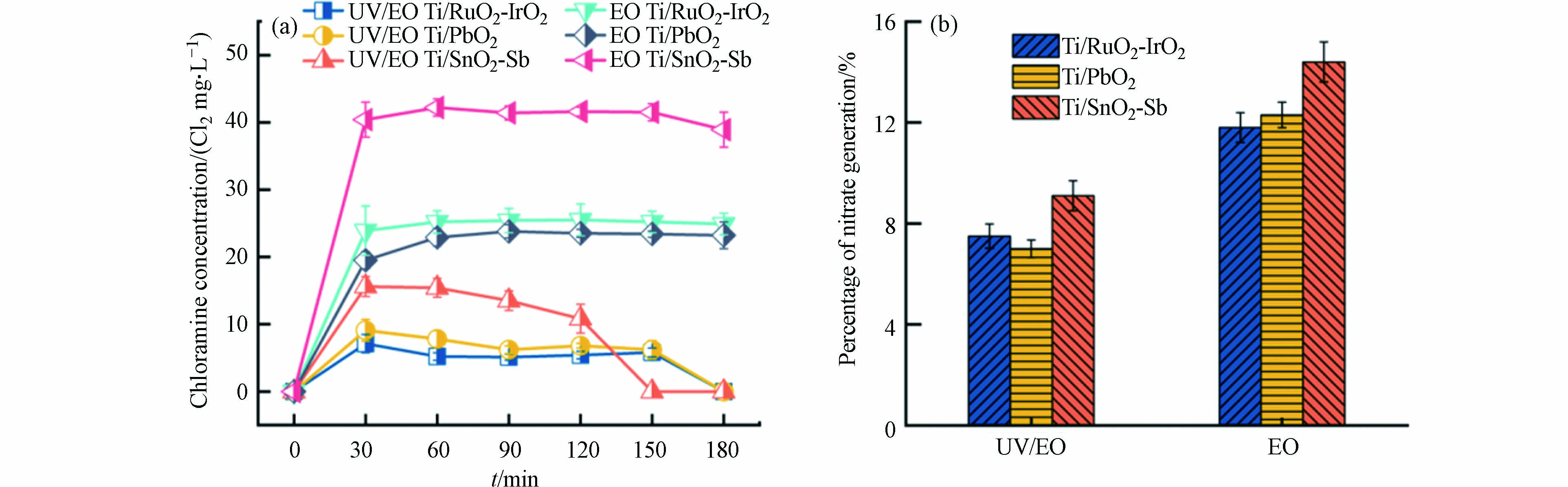
图 2 不同体系副产物生成对比(a)氯胺浓度和(b)硝酸盐生成的占比
Figure 2. Comparisons of by-product generation between different systems: (a) chloramine concentration and (b) percentage of NO3−-N generation

图 3 3种电极电化学表征及析氯性能比较:LSV曲线(a)Ti/RuO2-IrO2;(b)Ti/PbO2;(c)Ti/SnO2-Sb及(d)游离氯生成情况
Figure 3. Electrochemical characterization and comparison of chlorine evolution performance between three electrodes: LSV curves of (a) Ti/RuO2-IrO2, (b) Ti/PbO2, and (c) Ti/SnO2-Sb; and (d) free chlorine generation

图 4 不同体系中3种阳极EPR光谱比较(a)Ti/RuO2-IrO2;(b)Ti/PbO2;(c)Ti/SnO2-Sb以及掩蔽实验(d)Ti/RuO2-IrO2;(e)Ti/PbO2;(f)Ti/SnO2-Sb
Figure 4. Comparisons of EPR spectra of three anodes between different systems: (a) Ti/RuO2-IrO2; (b) Ti/PbO2; and (c) Ti/SnO2-Sb. Quenching experiments of (d) Ti/RuO2-IrO2; (e) Ti/PbO2; and (f) Ti/SnO2-Sb

图 5 不同体系3种电极处理实际废水对比:(a)金属冲洗水及(b)氨气吸收塔清洗水氨氮降解随时间变化;(c)金属冲洗水及(d)氨气吸收塔清洗水电化学能耗
Figure 5. Comparisons between different systems for real wastewater treatment: time courses of NH4+-N concentration in (a) metal washing and (b) ammonia-absorption-tower cleaning wastewater, and energy consumption for the treatment of (c) metal washing and (d) ammonia-absorption-tower cleaning wastewater

图 6 不同阳极(a)加速寿命实验结果及(b)去除1 kg NH4+-N所耗费的电极费用估算
Figure 6. Comparisons between different anodes for (a) the results of accelerated electrode life experiments and (b) estimated cost of anode in terms of 1 kg NH4+-N removal
表 1 真实废水水质参数
Table 1. Water quality characteristics of real wastewater
水样
EntrypH NH4+-N/
(mg·L−1)NO3−-N/
(mg·L−1)NO2−-N/
(mg·L−1)TOC/
(mg·L−1)Cl−/
(mg·L−1)金属冲洗废水
Metal rinsing wastewaters1.89 47.75 ± 0.97 0.45 ± 0.27 ND. 15.59 ± 1.97 561.89 ± 14.16 氨气吸塔清洗废水
Ammonia absorption tower cleaning wastewaters2.24 61.59 ± 2.17 0.23 ± 0.17 ND. 18.89 ± 2.24 712.46 ± 23.93  下载: 导出CSV
下载: 导出CSV
-
[1] YAN Z, DAI Z R, ZHENG W X, et al. Facile ammonium oxidation to nitrogen gas in acid wastewater by in situ photogenerated chlorine radicals [J]. Water Research, 2021, 205: 117678. doi: 10.1016/j.watres.2021.117678 [2] KEARNEY D, DUGAUGUEZ O, BEJAN D, et al. Electrochemical oxidation for denitrification of ammonia: A conceptual approach for remediation of ammonia in poultry barns [J]. ACS Sustainable Chemistry & Engineering, 2013, 1(1): 190-197. [3] WANG S, YE Z Y, TAGHIPOUR F. UV photoelectrochemical process for the synergistic degradation of total ammonia nitrogen (TAN) [J]. Journal of Cleaner Production, 2021, 289: 125645. doi: 10.1016/j.jclepro.2020.125645 [4] WANG R, SHU J C, CHEN M J, et al. An innovative method for fractionally removing high concentrations of Ni2+, PO43−, TP, COD, and NH4+-N from printed-circuit-board nickel plating wastewater [J]. Separation and Purification Technology, 2021, 260: 118241. doi: 10.1016/j.seppur.2020.118241 [5] SU B S, LIU Q, LIANG H L, et al. Simultaneous partial nitrification, anammox, and denitrification in an upflow microaerobic membrane bioreactor treating middle concentration of ammonia nitrogen wastewater with low COD/TN ratio [J]. Chemosphere, 2022, 295: 133832. doi: 10.1016/j.chemosphere.2022.133832 [6] 张道斌, 吕玉娟, 张晖. 化学沉淀法去除垃圾渗滤液中氨氮的试验研究 [J]. 环境化学, 2007, 26(1): 62-65. doi: 10.3321/j.issn:0254-6108.2007.01.015 ZHANG D B, LU Y J, ZHANG H. Ammonia-nitrogen removal from landfill leachate by chemical precipitation [J]. Environmental Chemistry, 2007, 26(1): 62-65(in Chinese). doi: 10.3321/j.issn:0254-6108.2007.01.015
[7] 张曦, 吴为中, 温东辉, 等. 氨氮在天然沸石上的吸附及解吸 [J]. 环境化学, 2003, 22(2): 166-171. doi: 10.3321/j.issn:0254-6108.2003.02.012 ZHANG X, WU W Z, WEN D H, et al. Adsorption and desorption of ammonia-nitrogen onto natural zeolite [J]. Environmental Chemistry, 2003, 22(2): 166-171(in Chinese). doi: 10.3321/j.issn:0254-6108.2003.02.012
[8] JAFVERT C T, VALENTINE R L. Reaction scheme for the chlorination of ammoniacal water [J]. Environmental Science & Technology, 1992, 26(3): 577-586. [9] KARRI R R, SAHU J N, CHIMMIRI V. Critical review of abatement of ammonia from wastewater [J]. Journal of Molecular Liquids, 2018, 261: 21-31. doi: 10.1016/j.molliq.2018.03.120 [10] SHIN Y U, YOO H Y, KIM S, et al. Sequential combination of electro-Fenton and electrochemical chlorination processes for the treatment of anaerobically-digested food wastewater [J]. Environmental Science & Technology, 2017, 51(18): 10700-10710. [11] KIM K W, KIM Y J, KIM I T, et al. Electrochemical conversion characteristics of ammonia to nitrogen [J]. Water Research, 2006, 40(7): 1431-1441. doi: 10.1016/j.watres.2006.01.042 [12] MOREIRA F C, BOAVENTURA R A R, BRILLAS E, et al. Electrochemical advanced oxidation processes: A review on their application to synthetic and real wastewaters [J]. Applied Catalysis B:Environmental, 2017, 202: 217-261. doi: 10.1016/j.apcatb.2016.08.037 [13] GARCIA-RODRIGUEZ O, MOUSSET E, OLVERA-VARGAS H, et al. Electrochemical treatment of highly concentrated wastewater: A review of experimental and modeling approaches from lab- to full-scale [J]. Critical Reviews in Environmental Science and Technology, 2022, 52(2): 240-309. doi: 10.1080/10643389.2020.1820428 [14] BUNCE N J, BEJAN D. Mechanism of electrochemical oxidation of ammonia [J]. Electrochimica Acta, 2011, 56(24): 8085-8093. doi: 10.1016/j.electacta.2011.07.078 [15] RADJENOVIC J, SEDLAK D L. Challenges and opportunities for electrochemical processes as next-generation technologies for the treatment of contaminated water [J]. Environmental Science & Technology, 2015, 49(19): 11292-11302. [16] LI L, LIU Y. Ammonia removal in electrochemical oxidation: Mechanism and pseudo-kinetics [J]. Journal of Hazardous Materials, 2009, 161(2-3): 1010-1016. doi: 10.1016/j.jhazmat.2008.04.047 [17] ZHENG W X, ZHU L Y, LIANG S, et al. Discovering the importance of ClO• in a coupled electrochemical system for the simultaneous removal of carbon and nitrogen from secondary coking wastewater effluent [J]. Environmental Science & Technology, 2020, 54(14): 9015-9024. [18] ZHANG Y, LI J H, BAI J, et al. Extremely efficient decomposition of ammonia N to N2 using ClO• from reactions of HO• and HOCl generated in situ on a novel bifacial photoelectroanode [J]. Environmental Science & Technology, 2019, 53(12): 6945-6953. [19] KUANG W J, YAN Z, CHEN J X, et al. A bipolar membrane-integrated electrochlorination process for highly efficient ammonium removal in mature landfill leachate: The importance of ClO• generation[J]. Environmental Science & Technology, 2022,10: 36240017. [20] KÉKEDY -NAGY L, ENGLISH L, ANARI Z, et al. Electrochemical nutrient removal from natural wastewater sources and its impact on water quality [J]. Water Research, 2022, 210: 118001. doi: 10.1016/j.watres.2021.118001 [21] MINAKATA D, KAMATH D, MAETZOLD S. Mechanistic insight into the reactivity of chlorine-derived radicals in the aqueous-phase UV–chlorine advanced oxidation process: Quantum mechanical calculations [J]. Environmental Science & Technology, 2017, 51(12): 6918-6926. [22] KAPAŁKA A, KATSAOUNIS A, MICHELS N L, et al. Ammonia oxidation to nitrogen mediated by electrogenerated active chlorine on Ti/PtOx-IrO2 [J]. Electrochemistry Communications, 2010, 12(9): 1203-1205. doi: 10.1016/j.elecom.2010.06.019 [23] LIN K N, ZHU Y, ZHANG Y B, et al. Determination of ammonia nitrogen in natural waters: Recent advances and applications [J]. Trends in Environmental Analytical Chemistry, 2019, 24: e00073. doi: 10.1016/j.teac.2019.e00073 [24] YANG Y, SHIN J, JASPER J T, et al. Multilayer heterojunction anodes for saline wastewater treatment: Design strategies and reactive species generation mechanisms [J]. Environmental Science & Technology, 2016, 50(16): 8780-8787. [25] LIU Y, TUCKERMAN M E. Protonic defects in hydrogen bonded liquids: Structure and dynamics in ammonia and comparison with water [J]. The Journal of Physical Chemistry B, 2001, 105(28): 6598-6610. doi: 10.1021/jp010008a [26] SHE L N, ZHAO G Q, MA T Y, et al. On the durability of iridium-based electrocatalysts toward the oxygen evolution reaction under acid environment [J]. Advanced Functional Materials, 2022, 32(5): 2108465. doi: 10.1002/adfm.202108465 [27] ZHU W J, HUANG Z H, ZHAO M T, et al. Hydrogen production by electrocatalysis using the reaction of acidic oxygen evolution: A review [J]. Environmental Chemistry Letters, 2022, 20(6): 3429-3452. doi: 10.1007/s10311-022-01454-5 [28] DEBORDE M, von GUNTEN U. Reactions of chlorine with inorganic and organic compounds during water treatment—Kinetics and mechanisms: A critical review [J]. Water Research, 2008, 42(1-2): 13-51. doi: 10.1016/j.watres.2007.07.025 [29] STANBURY D M. Mechanisms of advanced oxidation processes, the principle of detailed balancing, and specifics of the UV/chloramine process [J]. Environmental Science & Technology, 2020, 54(7): 4658-4663. [30] ZHOU S Q, WU Y T, ZHU S M, et al. Nitrogen conversion from ammonia to trichloronitromethane: Potential risk during UV/chlorine process [J]. Water Research, 2020, 172: 115508. doi: 10.1016/j.watres.2020.115508 [31] HA M R, THANGAVEL P, DANG N K, et al. High-performing atomic electrocatalyst for chlorine evolution reaction[J]. Small, 2023: 2300240. [32] HA H, JIN K, PARK S, et al. Highly selective active chlorine generation electrocatalyzed by Co3O4 nanoparticles: Mechanistic investigation through in situ electrokinetic and spectroscopic analyses [J]. The Journal of Physical Chemistry Letters, 2019, 10(6): 1226-1233. doi: 10.1021/acs.jpclett.9b00547 [33] GENDEL Y, LAHAV O. Revealing the mechanism of indirect ammonia electrooxidation [J]. Electrochimica Acta, 2012, 63: 209-219. doi: 10.1016/j.electacta.2011.12.092 [34] ZHANG S C, CHEN X, DU S W, et al. Facile synthesis of highly active Ti/Sb-SnO2 electrode by sol-gel spinning technique for landfill leachate treatment [J]. Water Science and Technology, 2021, 84(6): 1366-1378. doi: 10.2166/wst.2021.336 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 2634
- HTML全文浏览数: 2634
- PDF下载数: 110
- 施引文献: 0
