

-
六氯丁二烯 (Hexachlorobutadiene, HCBD) 是一无自然来源的新型氯代持久性有机污染物 ,具有长距离迁移性,且对生物体具有生殖、遗传和潜在的致癌毒性[1],于2015年被列入《关于持久性有机污染物的斯德哥尔摩公约》. 虽然HCBD已被禁止生产和使用,但氯代烃(例如,三氯乙烯和四氯乙烯)合成过程中的非故意排放仍是目前我国HCBD的主要来源[2,3]. 研究发现我国 1992年—2016 年间HCBD 的历史非故意排放年排放量从 60.8 Mt·a−1增加到
2871.5 Mt·a−1 [4],目前,HCBD在水[5,6]、土壤[7]、大气[8]和沉积物,甚至南极冰芯中等环境介质中均被检出[9],HCBD在多介质环境中的污染问题引起了国家的高度重视. 在2022年12月,HCBD被列入于2023年3月1日正式施行的《重点管控新污染物清单(2023年版)》[10]. 因此,HCBD污染的高效控制和削减刻不容缓.目前对于环境中HCBD处置方法研究主要聚焦于加氢脱氯研究,尤其是基于微生物介导法实现HCBD的还原性脱氯. 然而加氢脱氯并不能实现HCBD的完全降解[5,11,12],脱氯中间产物仍存在二次污染的隐患. 热催化降解技术因其能在相对较低温度下破坏污染物结构,使其转化为相对无害的CO2、H2O、HCl等低毒或无毒产物,广泛应用于氯代持久性有机污染物(persistent organic pollutants, POPs)的处置[13]. 在氯代POPs的催化降解研究中,贵金属催化剂具有优异的低温催化活性,但存在成本高、与氯原子结合易失活等问题. 因此,价格低廉且具备活性优势的复合金属氧化物引起了研究者们广泛的关注[14]. 其中,锰基莫来石SmMn2O5 (AB2O5) 存在金属—氧多面体骨架,兼具稳定性与高活性[15]. 已有研究表明SmMn2O5中B 位锰原子是其主要的活性位点,含有两种不同的晶体场:以六个氧原子配位的八面体形式存在的Mn4+和以五个氧原子配位的正四面体形式存在的Mn3+、Mn3+和 Mn4+金属离子共存,形成了独特Mn3+—O—Mn4+二聚体,提供了丰富的氧空位和良好的氧迁移性,保证了锰基莫来石SmMn2O5催化剂的高活性[16 − 19]. 同时,Mn3+和Mn4+的混合存在有利于Mn—O键的形成与断裂,保证了其结构的稳定性[20]. AB2O5类化合物 (如:SmMn2O5) 中A位元素不会直接参与催化反应,但A位元素的改变会导致电子构型发生变化,对催化剂的活性产生一定影响[21,22]. 为了进一步提升锰基莫来石SmMn2O5的催化性能,目前较多研究通过对A位元素掺杂和取代其他金属元素来增加活性比表面积或调整氧活化能力,但其机制因掺杂元素不同而有所差异. Feng[23]等在 SmMn2O5的A位引入不同比例的La,发现La3+部分取代Sm3+会导致催化剂形成结构缺陷和氧空位,进而有效地改变反应中间产物的稳定性,促进 NO 的氧化. Yang[24] 等研究发现A位Ba的引入,会导致中间产物亚硝酸盐物种失稳,促进NO的氧化;而Sr和La的引入提高了O2的解离能力,使SmMn2O5在较宽的温度区间均有较强的反应活性. 相比而言,通过有效的策略来调节Mn位点以提高氧化还原能力和增加活性位点较少被研究. Yang等通过酸刻蚀改变Mn—O的共价性,促进Mn和O之间电荷转移,增加了活性Mn位点的暴露量,提高了催化剂的催化性能[16,25],然而B位Mn可变价的过渡金属作为主要的活性中心,其直接调控对催化剂结构与性能的影响研究较少. Shen[17] 等研究了B位Cu掺杂的SmMn2O5莫来石型催化剂并对其进行了酸刻蚀,发现高活性的Cu2+-O-Mn4+位点与酸刻蚀协同作用增强了表面晶格氧的活性,促进了中间物种的有效转化. B位点直接调控对催化剂性能及结构的亟需进一步探究.
本研究从Mn—Mn二聚体活性位点出发,基于SmMn2O5开展B位元素调控,制备系列B位不同Co掺杂占比Sm(Mn1-xCox)2O5 (0≤x≤0.5)莫来石材料,研究系列调控材料对HCBD的催化降解活性和反应动力学,探究B位Co元素不同掺杂占比对催化剂晶相、形貌、活性氧物种、酸性位点等结构特征的影响,明晰HCBD在催化剂表面的界面反应过程,阐明B位Co掺杂对莫来石催化性能的调控机制,揭示HCBD的相关热催化降解机制,为HCBD控制研究技术提供思路与理论依据.
-
硝酸钐(III),六水合物(分析纯),四甲基氢氧化铵溶液,硝酸锰溶液(wt%,50%分析纯),上海麦克林生化科技有限公司;Pluronic F-127,六氯丁二烯(96%标准品),Sigma-Aldrich;30% 过氧化氢(优级纯),沪试试剂;无水硫酸钠(优级纯),科密欧试剂;正己烷(色谱纯),Fisher Chemica;六氯丁二烯气体(20 mg·m−3标准气体),北京兆格气体科技有限公司;硅烷化玻璃棉,上海安谱实验科技有限公司;高纯氮气(99.999%),高纯氦气(99.999%)北京兆格气体科技有限公司;高纯氧气(99.999%)海科元昌实用气体有限公司;氨气/氮气(0.105%)华元气体化工有限公司.
-
制备Sm(Mn1-xCox)2O5 (x = 0、0.1、0.2、0.3、0.4、 0.5) 系列催化剂,具体制备方法如下:以理论制备1 g Sm(Mn1-xCox)2O5催化剂为计算基准,按比例称取六水合硝酸钐、硝酸钴置于100 mL 烧杯中,随后加入一定体积硝酸锰溶液,加入40 mL 超纯水搅拌溶解. 称取0.04 g 表面活性剂Pluronic F-127加入正在搅拌的混合液中,加入四甲基氢氧化铵调节溶液pH值至11,随后加入适量体积过氧化氢溶液(理论计算量为超过 B 位元素质量 40%),常温搅拌2 h. 搅拌后的溶液转移至50 mL离心管中,在
3800 r·min−1转速下离心 5 min,随后将沉淀部分放置在100 ℃ 鼓风干燥箱中烘 12 h,得到材料前驱体.将烘干后的材料放入玛瑙研钵中,研磨后放入氧化铝坩埚中,在500 ℃ 煅烧 8 h,800 ℃煅烧8 h,升温速率为5 ℃·min−1,冷却后取出,研磨制得Sm(Mn1-xCox)2O5 (x = 0、0.1、0.2、0.3、0.4、 0.5)系列催化剂粉末.
-
在密闭体系中热能量场作用下,模拟化合物HCBD的催化降解. 具体实验步骤如下:密闭体系为定制玻璃管(外径8 mm,内径 5 mm,长度为12 cm),取适量硅烷化玻璃棉置于玻璃管适当位置. 将1 μL HCBD用微量进样针打至玻璃棉上,称取100 mg Sm(Mn1-xCox)2O5 (0≤x≤0.5)催化剂转移至负载HCBD的玻璃棉上,随后用酒精喷灯将玻璃管另一端封闭,制成体积约为1.6 mL密闭样品管. 置于已预热为300 ℃的马弗炉内. 准备三组平行样品,同时设置用微量进样针打至玻璃棉上1 μL HCBD后直接封管的对比样品三组. 将三组样品管与三组对比管同时放入预热至300 ℃ 的马弗炉中,迅速关闭. 计时5 min,迅速用坩埚钳取出放置样品管的坩埚,冷却至室温. 以相同方式分别准备10 min、20 min、40 min和60 min的样品及0 min的空白样品.
-
降解后的反应体系中HCBD的检测方式如下:用硫酸纸 (14 × 14 cm) 将样品管包裹严实后,置于干净铝箔上,用铁锤小心敲碎封管上端,将敲碎后的玻璃管碎屑和管内反应后物质一并转移至100 mL烧杯中. 向烧杯中加入20 mL正己烷,采用超声萃取5 min后取出静置. 用无水硫酸钠填充砂板小柱,将静置后的上清液转移到小柱中. 重复萃取3次. 将萃取液混合并定容至100 mL棕色容量瓶中,待测. 回收率实验为不加热的三组用微量进样针打至玻璃棉上1 μL HCBD 后直接封管的空白样品管. 采用7890B GC—7000A QQQ三重四极杆气质联用对未完全降解的 HCBD 进行定量分析. 仪器的具体参数如下:载气为高纯氦气,流速为 1.5 mL·min−1,离子源为电子轰击电离源(EI),色谱柱为 DB—5 MS 毛细管柱(30 m×250 μm,膜厚0.25 μm),离子源和进样口温度分别为 150 ℃和 250 ℃,升温程序为:初始柱温50 ℃,保持 1 min,随后以10 ℃· min−1 的升温速率升至 100 ℃,保持 1 min,然后再以 5 ℃·min−1 的升温速率升至 120 ℃,保持 1 min;不分流进样,每次进样体积为 1 μL;采用选择离子模式对产物进行检测,检测离子 m/z 为 223、225、231和233.
活性评价按照公式(1)来计算:
式中,RHCBD 表示样品管中的HCBD剩余的摩尔量,mol;IHCBD 表示HCBD的初始摩尔量,mol.
-
采用X射线粉末衍射(X—ray Diffraction , XRD)对催化剂的晶体结构进行表征,仪器为X'Pert PRO功率衍射仪,PANalytical. 将适量的样品分散在载玻片刻蚀凹槽处,用洁净盖玻片压实,保证材料表面平整,随后进行 XRD 测试. 激发光源为 Cu Kα 射线 (λ=
1.5418 Å),定管电压为40 kV,管电流为40 mA,扫描范围 (2θ) 为 5°—90°,扫描速度为5 °·min−1. 采用共焦显微拉曼光谱仪 (inVia Raman microscope, Renishaw),将适量样品分散在载玻片上,用洁净盖玻片压平表面,随后进行测试. 激光波长采用523 nm,Extended模式,范围为200—1200 nm,栅格为:2400 line·mm−1, 激光功率为10% ,曝光时间为10 s. 采用场发射扫描电子显微镜(Scanning Electron Microscope, SEM,日本Hitachi Regulus8100 )获取催化剂表面微观形貌特征,取微量催化剂直接粘到导电胶上,并使用 Quorum SC7620 溅射镀膜仪喷金45 s,喷金为10 mA;随后使用Hitachi Regulus8100 扫描电子显微镜拍摄样品形貌、能谱mapping等测试,形貌拍摄时加速电压为3 kV,能谱mapping拍摄时加速电压为20 kV,探测器为SE2二次电子探测器. 使用Micromeritics ASAP2460 表面积和孔隙度分析仪,在77 K下通过Brunner-Emmet-Teller (BET) 分析来确定比表面积. 在进行N2 物理吸附之前,对准备好的催化剂在 300 ℃的真空条件下进行6 h 脱气预处理.使用X 射线光电子能谱(X—Ray Photoelectron Spectroscopy, XPS, Thermo Scientific K—Alpha)分析催化剂表面的元素组成与化学价态,取适量催化剂压片后,贴于样品盘上,将样品放进Thermo Scientific K—Alpha XPS仪器样品室中,在样品室的压力小于2.0×10−7 mbar时,将样品送入分析室,光斑大小为400 μm,工作电压12 kV,灯丝电流6 mA. 扫描通能为50 eV,步长 0.1 eV. 通过设置表面不定碳(C 1s)的结合能为 284.84 eV,校准了催化剂的结合能刻度. 使用傅里叶变换红外光谱(Fourier Transform infrared spectroscopy, FT—IR,德国布鲁克,INVENIO—R)在300 ℃,50 mL·min−1N2氛围预处理30 min,降温至室温后稳定15 min,扫描背景. 随后在50 mL·min−1 NH3氛围下(0.105%,N2作为平衡气)吸附30 min,50 mL·min−1 N2氛围脱附30 min,扫描脱附图谱.
氢气—程序升温还原(Hydrogen Temperature-Programmed Reduction,H2—TPR,美国Micromeritics AutoChem II
2920 ),称取100 mg样品置于U型石英管中,以10 ℃·min−1从室温程序升温至300℃干燥预处理,He 气流 (50 mL·min−1) 吹扫1 h,冷却至50 ℃,通入10% H2/Ar混合气 (50 mL·min−1) 0.5h待基线稳定后,样品在10% H2/Ar气流中以10 ℃·min−1 的升温速率升至800 ℃脱附,用TCD检测还原气体.通过气相色谱—火焰例子检测器/质谱检测器 (Gas chromatography-Flame Ionization Detector/Mass Spectrometer,GC—FID/MS) 联用系统进行了定量分析. 称取100 mg待测样品,在300 ℃持续通入40 mL·min−1空气,待稳定后注射1 μL六氯丁二烯进行反应,测试反应后尾气产物. 具体测试流程如下:通过三级制冷方法用液氮对气体进行预浓缩,样品气体经过前两级冷阱去除N2、CO2、O2、H2O等干扰物. 随后将预浓缩的气体在毛细管柱子顶部热解吸,并通过GC—FID分析尾气中乙烯、乙炔和乙烷的浓度. 最后,过HP-1毛细管柱 (60 m × 0.32 mm × 1 μm,100% 二甲基聚硅氧烷,Agilent,美国) 对所采集样品中其他产物进行GC—MS (Agilent7890AGC,Agilent5977B MS) 定量分析. GC的箱体温度首先在35 ℃下保持5 min,然后以5 °C·min−1的速率升高至200 ℃,最后在200 °C下保持8 min. 使用选择离子监测(SIM)模式在质谱中采用电子轰击(EI)离子源,离子源的温度设置为230 ℃.
-
Sm(Mn1-xCox)2O5 (x = 0,0.1,0.2,0.3,0.4, 0.5) 系列催化剂对HCBD的降解活性评价结果如图1所示,每一组活性实验结果均进行了重复实验,其标准差在图中以误差棒的形式呈现. SmMn2O5对HCBD的60 min降解率为58%. 当B位Co的掺杂占比为0.1时,催化剂对HCBD的降解率稍有提高,略大于60%;当Co的掺杂占比为0.2时,在60 min反应时间内,HCBD的降解效率达到了约66%;当掺杂量进一步增加,即Co掺杂占比为0.3时,HCBD降解效率略有下降;随着Co掺杂占比的进一步增多,x = 0.4时,降解率明显下降;当B位元素Mn、Co占比为1:1时,HCBD的降解效率又有略微的回升. 莫来石的降解效率与B位Co的掺杂占比呈现出火山型波动式变化,并非简单的线性关系. Sm(Mn0.8Co0.2)2O5表现出较为优异的降解活性.
根据HCBD随着反应时间变化的剩余量,探究了催化剂对其的反应动力学. 分析发现HCBD在Sm(Mn1-xCox)2O5 (0≤x≤0.5)上的反应均符合准一级动力学,其动力学方程如式(2):
其中,RHCBD 表示样品管中的HCBD剩余的摩尔量,mol;IHCBD表示HCBD的初始摩尔量,mol. Kobs为一级动力学反应速率常数,单位为min−1;t为反应时间,单位为min.
如表1所示,对六种催化剂材料来说,所得HCBD的ln[(RHCBD)/(IHCBD)]与反应时间存在线性关系,其中Sm(Mn0.8Co0.2)2O5、Sm(Mn0.7Co0.3)2O5和SmMn2O5催化降解HCBD的准一级动力学拟合见图2. 拟合所得HCBD在SmMn2O5、Sm(Mn0.9Co0.1)2O5、Sm(Mn0.8Co0.2)2O5、Sm(Mn0.7Co0.3)2O5、Sm(Mn0.6Co0.4)2O5、Sm(Mn0.5Co0.5)2O5材料上的准一级反应动力学常数分别为
0.0142 min−1(R2=0.99)、0.0149 min−1(R2=0.98)、0.019 min−1(R2=0.99)和0.0173 min−1(R2=0.99)、0.0146 min−1(R2=0.99)、0.0156 min−1 (R2=0.96). 与钐锰莫来石相比,Sm(Mn1-xCox)2O5 (0≤x≤0.5)对HCBD的降解反应速率有所提升,尤其是Sm(Mn0.8Co0.2)2O5,对HCBD的降解反应速率最快. -
为了探究B位Co不同掺杂占比调控对钐锰莫来石活性的影响,本研究首先利用XRD、Raman表征分析了Sm(Mn1-xCox)2O5(0≤x≤0.5)系列催化剂进的晶相组成,同时利用SEM—EDS观察其微观形貌与表面元素分布情况,采用 BET分析了系列催化剂的比表面积与孔径等参数. 其中SmMn2O5 及Sm(Mn1-xCox)2O5(0<x≤0.5)系列催化剂 XRD 表征结果见图3. 少量Co的引入并未改变莫来石晶相. Mn3+和Mn4+的离子半径分别为0.785 Å和0.67Å,而Co2+、Co3+的离子半径分别为0.74 Å和0.63 Å (Shannon Radii (ic.ac.uk)),表明Co2+更有可能占据Mn3+位点,而Co3+则有可能占据Mn4+位点,随后形成Co2+—O—Mn4+和Mn3+—O—Co3+[17]. 随着Co掺杂占比的增大,当x = 0.2时,Sm(Mn0.8Co0.2)2O5出现了微量的钙钛矿型氧化物SmMnO3相(PDF#00—025—0747). 伴随着Co在B位掺杂占比的进一步增加,催化剂中SmMnO3晶相特征峰相对强度逐渐增强,同时SmMn2O5晶相特征峰相对强度逐渐减弱. 当Co的掺杂占比为0.4时,少量的SmCoO3(PDF#00—025—
1071 )生成,其最强特征峰33.74°与位于33.8°属于SmMn2O5特征峰重合,导致此处峰强略增强. 当x = 0.5时,SmMn2O5相与SmMnO3钙钛矿相二者特征峰最强峰相对强度相当.为了考察B位Co掺杂占比对各物相生成的影响,对XRD图谱进行了精修,根据精细图对样品中的不同物相含量进行计算,结果如图4. 钙钛矿型氧化物SmMnO3晶相占比随着Co掺入占比增大而持续增加,由Sm(Mn0.8Co0.2)2O5中的13.1%增加至Sm(Mn0.6Co0.4)2O5中的37.3%,表明Co进入SmMn2O5中取代B位Mn,同时生成了新的物相SmMnO3. Sm(Mn0.5Co0.5)2O5中SmMnO3占比为37.7%,相对于Sm(Mn0.6Co0.4)2O5中37.3%中仅微量增加,表明Co在B位中对Mn的取代接近饱和,在Co掺入占比为0.4时,部分Co形成少量新物相SmCoO3,Co掺杂占比的进一步增加则会生成更多SmCoO3晶相.
为了进一步明确Co掺杂后SmMn2O5结构变化,对SmMn2O5及Sm(Mn1-xCox)2O5(0<x≤0.5)系列催化剂进行了Raman光谱表征,结果如图4所示. SmMn2O5在208 cm−1处的峰为Mn—Mn特征振动峰,606 cm−1和617 cm−1的峰归属为Mn4+—O的特征振动峰,671 cm−1的峰为Mn3+—O的特征振动峰[18,19]. 当Co开始掺入时,在x = 0.1—0.3,随着Co掺杂占比的增加,208 cm−1(Mn—Mn) 与671cm−1(Mn3+—O)处的特征峰明显逐渐变窄,说明Mn—Mn中有其他元素掺入取代部分Mn3+[17],表明Co在B位的调控以形成Co2+—O—Mn4+为主. Sm(Mn0.9Co0.1)2O5、Sm(Mn0.8Co0.2)2O5相对于SmMn2O5整体发生了红移,其中Sm(Mn0.9Co0.1)2O5的617 cm−1(Mn4+—O)和671 cm−1 (Mn3+—O) 均发生红移,而Sm(Mn0.8Co0.2)2O5主要为671 cm−1 (Mn3+—O) 发生红移. 表明相比于其他掺杂配比,适量Co2+—O—Mn4+的存在可能促进金属氧键的断裂,提高氧的移动性并促使活性氧物种的生成,进而有利于催化降解活性的提升. 在 x = 0.3—0.5时,相较于208 cm−1(Mn—Mn),606 cm−1处峰相对强度明显增强,基于XRD表征结果,这可能是因为SmMnO3在607 cm−1处存在由于正交钙钛矿结构中B2g(1)振动产生的峰[26],与SmMn2O5在606 cm−1处存在的峰重合. 因此,随着Co掺杂量的进一步增加,SmMnO3晶相生成,使607 cm−1处相对峰强明显增加,同时208 cm−1(Mn—Mn)峰的相对峰强显著下降,与XRD数据分析结果一致.
明确催化剂结构后,我们对Sm(Mn1-xCox)2O5(0≤x≤0.5)系列催化剂的微观形貌进行了SEM表征,采用EDS mapping模式分析系列催化剂表面元素分布,结果如图5所示. SmMn2O5莫来石呈现不规则纳米棒状结构,这些纳米棒可能是由于晶粒沿着特定晶面取向生长所致[27]. 随着Co的掺杂,不规则短棒逐渐变为六棱柱状,同时表面出现不同于规则短棒状结构的其他物质,结合XRD数据分析推断可能为SmMnO3,与之前的研究结果一致[28]. SmMn2O5 及Sm(Mn1-xCox)2O5(0<x≤0.5)系列催化剂元素EDS mapping结果显示,Sm、Mn、Co以及O均在材料表面分布均匀. 系列催化剂比表面积分析结果如表2所示. 与SmMn2O5催化剂相比,B位Co掺杂后,系列材料的比表面积、平均孔径和孔体积均有明显降低.
Co掺杂比例为0.1时,Sm(Mn0.9Co0.1)2O5比表面积为11.16 m2·g−1. 当x = 0.1—0.3时,随着Co掺杂比例的增加,催化剂的比表面积相差不大,但其孔体积和平均孔径均略减小. 结合XRD分析结果,在x =0.1—0.3时仅出现了极少量的SmMnO3. 当 x = 0.3—0.5时,随着Co掺杂比例的增加,催化剂比表面积减小,但其孔体积和平均孔径均增大,可能是由于SmMnO3晶相的逐渐增强. 结合催化剂的活性表现分析,Co掺杂后的材料整体比表面积低于SmMn2O5,然而掺杂后材料整体的活性相比于钐锰莫来石均有不同程度的提高. 这表明比表面积并不是催化性能提升的主要原因,Co引入形成的Co2+-O-Mn4+及钙钛矿的形成导致催化剂表面结构配位变化对性能的影响可能更为关键.
-
结合活性评价数据结果发现,SmMn2O5莫来石在B位掺杂少量Co能够在一定程度上提高催化剂对HCBD的降解效率. 当Co的掺杂量增多,材料的晶相逐渐发生变化. 系列材料的主要晶相仍是SmMn2O5莫来石相,部分SmMnO3与SmCoO3的形成以及Co对B位Mn的取代改变了原本B位Mn元素的配位环境,进而导致催化剂对HCBD的降解活性发生变化. 然而,催化活性的变化与Co掺杂量并非为简单的线性关系,为进一步探究材料晶相变化与B位配位改变对催化活性的影响,利用XPS和NH3—FTIR分析Sm(Mn1-xCox)2O5(0≤x≤0.5)系列样品表面化学性质.
对Sm(Mn1-xCox)2O5(0≤x≤0.5)系列样品表面关键元素价态和配位环境进行了XPS分析,结果如图6所示. 图6 a为Mn 2p谱图,包含两种价态的Mn,其中结合能在642.04 eV—642.08 eV 处为Mn4+,640.70—640.73 eV处为Mn3+[29,30]. 分峰后根据相应峰面积计算Mn4+在所有Mn物种中的占比,可以发现随着Co掺杂比例的增加,Mn4+的占比增大,说明Co的掺入影响了锰原本的化学环境. 由XRD和Raman表征结果可知,Co在B位的调控主要为Co2+取代Mn3+,部分Mn3+—O—Mn4+转化形成Co2+—O—Mn4+,导致了Mn4+比例的升高.
从图6b Co 2p谱图可以看出,包含两种价态的Co,其中结合能在779.2—779.78 eV 处为 Co3+, 781.14—781.22 eV 处为Co2+[31,32]. 分峰后根据相应峰面积计算Co2+占比,在x = 0.1—0.3时,随着Co掺杂占比的增加,Co2+的占比逐渐增大. 在 x = 0.3—0.5时,随着Co掺杂占比的进一步增加,Co2+的占比减少,这可能是由于大部分Co以Co2+的形式掺杂到莫来石中,导致Co2+占比增大. 上述结果进一步证实了Co2+ 部分取代Mn3+—O—Mn4+中的Mn3+,形成了Co2+—O—Mn4+,改变了原本的Mn的配位环境. 而在 x = 0.4—0.5时,B位Co对Mn的取代达到饱和,相较于Sm(Mn0.6Co0.4)2O5,Sm(Mn0.5Co0.5)2O5中SmMnO3含量仅微量增加,过量的Co形成了SmCoO3钙钛矿,进而导致Sm(Mn0.6Co0.4)2O5和Sm(Mn0.5Co0.5)2O5中Co2+的占比相较于Sm(Mn0.7 Co0.3)2O5明显降低.
图6 c为O 1s谱图,结合能在528.56 eV—528.7 eV为晶格氧(Olatt : O2- ); 结合能在531.01—531.15 eV为表面吸附氧(Oads : O− 、O22-等);结合能在533.4 eV 的为表面吸附的水分子[33]. 其中表面吸附氧被广泛认为在氧化还原反应中具有更高的活性. 因此,我们对分峰后的氧物种峰面积进行计算,探究了系列催化剂中Oads/Ototal的分布规律. 发现当Co掺入占比为0.1时,Sm(Mn0.9Co0.1)2O5的Oads在所有氧物种中的占比变化不大. 而当x = 0.2时,Sm(Mn0.8Co0.2)2O5中Oads的占比明显变大,为0.5027. 而后随着Co的掺入比例的进一步增加,Oads占比略下降.
结合Mn 2p、Co 2p和O 1s的数据分析可得,适量Co的掺入,改变了SmMn2O5原本B位Mn元素的化学环境. Co2+对Mn3+的取代,使得SmMn2O5中Mn3+—O—Mn4+部分转化为Co2+—O—Mn4+,进而削弱金属氧键,促进活性氧的移动性,增强活性氧物种含量,提高了催化剂对HCBD的降解效率. 尤其是B位Co掺入量为0.2时,催化剂活性最好. 但当Co引入量较大时,莫来石虽为主导晶相,其他杂相的存在会降低活性氧物种占比,不利于催化活性的发生.
为探究系列催化剂的氧化还原性能,对Sm(Mn1-xCox)2O5(0≤x≤0.5)进行了H2—TPR表征,结果如图7所示. SmMn2O5低温区段的还原峰对应为Mn4+到Mn3+的还原,高温区段的特征峰归因于Mn3+到Mn2+的还原[34 − 36].
随着Co掺杂占比的增加,H2—TPR的两个特征峰逐渐向低温方向移动,表明Co掺杂提高了系列催化剂的氧化还原性. 同时,Co的掺杂导致了新的还原峰产生,尤其是Sm(Mn0.7Co0.3)2O5和Sm(Mn0.6Co0.4)2O5催化剂,530—560 °C低温区段的还原峰可能与Co3+到Co2+的还原及Mn4+到Mn3+的还原有关,而610 °C左右高温区段还原峰则归属为Co2+到Co0还原与Mn3+到Mn2+还原[14,36 − 39]. 根据各催化剂还原峰的温度分布,Sm(Mn0.7Co0.3)2O5、Sm(Mn0.6Co0.4)2O5和Sm(Mn0.5Co0.5)2O5的氧化还原活性高于Sm(Mn0.8Co0.2)2O5、Sm(Mn0.9Co0.1)2O5和SmMn2O5. 结合催化剂活性数据,5 min短时间内系列催化剂对HCBD的催化性能与上述氧化还原性能趋势分布相一致. 但随着反应时间的延长,Sm(Mn0.8Co0.2)2O5的催化活性逐渐增强,在60 min时对HCBD的降解效率达到最高. 这可能是由于适量Co的引入可延迟H2与催化剂表面活性锰离子的有效接触,进而延迟了氯物种对活性位点的氯化作用,而过强的氧化还原性导致催化剂更容易发生氯中毒[34],由此导致Sm(Mn0.6Co0.4)2O5 和Sm(Mn0.5Co0.5)2O5对HCBD的催化活性下降. 为了进一步研究催化剂表面的氯残留,我们利用XPS对催化反应后材料表面氯物种含量进行了分析(表3). 与较强氧化还原性的Sm(Mn0.6Co0.4)2O5 和Sm(Mn0.5Co0.5)2O5相比,Sm(Mn0.8Co0.2)2O5表面氯原子百分含量最低,为7.03%. 上述结果进一步证实了适当氧化还原性在氯代有机污染物催化降解过程中的关键作用.
在氯代有机物热催化降解过程中,酸性位点对于C—Cl键的吸附解离和Cl物种的脱附也发挥了重要作用[40,41]. 利用NH3—FTIR对Sm(Mn1-xCox)2O5(0≤x≤0.5)系列催化剂表面酸性位点进行了表征,结果图8所示.
3100 cm−1—2700 cm−1(B1)存在由NH4+的不对称和对称伸缩振动产生的较宽的弱吸收区域,1455 cm−1(B2)和1390 cm−1(B3)存在NH4+的不对称变形振动产生的吸收峰,表明催化剂表面存在Brønsted酸位. 此外,也存在氨分子在材料表面Lewis酸位点的吸收峰:在3400 —3100 cm−1 (L1)区域存在较宽的由于NH、NH2和NH3伸缩振动产生的较强吸收峰,1668 cm−1(L2)处为NH3的不对称变形振动产生的吸收峰,1327 cm−1(L3)处的吸收峰为NH3的对称变形振动产生,在970 cm−1(L4)处存在由于NH2弯曲振动产生的吸收峰[42]. 当B位Co的掺杂占比为0.1时,L3略有增加,但整体L酸和B酸变化不明显. 而当Co继续掺入时,催化剂表面的L酸和B酸位点均变丰富. 其中Sm(Mn0.8Co0.2)2O5、Sm(Mn0.7Co0.3)2O5中L1、L2、L3、B1、B2和B3峰强相对于SmMn2O5本身明显增大. 而随着Co掺入占比进一步增大,Sm(Mn0.6Co0.4)2O5和Sm(Mn0.5Co0.5)2O5中虽然仍有较多的L2和B3,但L1、B1和B2则有一定程度的减少. 在氯代有机物热催化降解过程中,酸性位点对于C-Cl键的吸附解离和Cl物种的脱附发挥了重要作用[40,41]. 一般认为,在氯代POPs催化降解过程中,Cl会优先吸附在Lewis酸位点,并且C—C和C—Cl键在Lewis酸位点发生裂解[43]. 因此,较高的Lewis酸度有利于提高催化剂对HCBD的吸附和矿化. Brønsted酸位点通过提供氢物种与催化剂表面的Cl物种生成HCl,有助于氯代产物的脱附,提高了催化剂的抗氯中毒性能[41]. 因此Sm(Mn0.8Co0.2)2O5中由于适量Co的引入,调控Co2+—O—Mn4+的形成,不仅增多了活性氧物种含量,还具有适度的氧化还原性能及较多的Lewis和Brønsted酸位点分布,进而在HCBD降解过程中表现出优于钐锰莫来石本身的催化活性. -
为了进一步探究B位调控钐锰莫来石催化剂在降解HCBD过程中的界面反应过程,本研究利用原位漫反射傅里叶变换红外光谱对不同温度下的界面过程开展原位表征. 然而,性能较为优异的Sm(Mn0.8Co0.2)2O5催化反应体系的原位红外谱图结果并不明显,这可能是由于Sm(Mn0.8Co0.2)2O5材料的物理化学性质影响红外光的穿透和反射,进而大幅降低了光谱峰的明显度. 对比SmMn2O5和Sm(Mn0.7Co0.3)2O5催化剂表面HCBD的界面反应(图9),发现二者中间产物特征峰类似,表明系列催化剂对HCBD的降解路径较为一致. 因此,选取Sm(Mn0.7Co0.3)2O5来进一步分析HCBD在B位Co引入钐锰莫来石催化剂上的界面反应路径. 如图11所示,998 cm−1处为HCBD中氯化亚甲基基团面内和面外振动产生的特征峰,
1214 cm−1处峰归属为HCBD中C—C键伸缩振动,1632 cm−1处峰归属为HCBD中C=C键伸缩振动[40,44](organchem.csdb.cn).1137 cm−1 为C—O—C(酯)对称伸缩或者C—O—C(醚)反对称伸缩振动产生的峰,而1076 cm−1为C—O 伸缩振动产生的峰[11]. 在Sm(Mn0.3Co0.7)2O5催化反应体系,50 ℃时在1632 cm−1处有HCBD微弱的吸附峰,当升温至100 ℃时,催化剂表面出现明显的HCBD吸附峰和中间产物峰;随着温度的继续升高,材料表面HCBD特征峰逐渐消失,中间产物特征峰也显著降低,说明随着温度的升高,吸附的HCBD和产生的中间产物逐渐被降解. 相比而言,SmMn2O5催化反应体系中在50 ℃出现了较为明显的HCBD吸附峰,随着温度的升高,HCBD吸附峰与中间产物特征峰也在逐渐降低,但在300 ℃ 反应温度下仍有一定量的母体污染物和中间产物. 这可能是由于Sm(Mn0.7Co0.3)2O5相较于SmMn2O5表面有更丰富的L酸和B酸位点,反应温度的升高导致表面氯物种脱附速率加快. 上述结果与活性评价结果相一致.为了探究系列催化剂对HCBD的降解机理,基于Sm(Mn1-xCox)2O5(0≤x≤0.5)中活性最优催化剂Sm(Mn0.8Co0.2)2O5,利用固定床微型反应装置与气相色谱-火焰例子检测器/质谱检测 (GC-FID/MS) 联用系统对产物进行分析,结果如图10所示. 二氯甲烷、甲醛、丙酮等是HCBD的主要降解产物,同时尾气中还检测到微量烷烃、烯烃、酮和其他小分子氯代烯烃等物质.
结合B位引入Co对SmMn2O5催化剂的结构调控和界面反应过程探究,提出了Co调控钐锰莫来石对HCBD的催化降解机制,如图11所示. B 位适量的Co的掺杂,一方面能够改变Mn 的配位环境,导致部分Co2+取代Mn3+形成Co2+—O—Mn4+,拉伸金属氧键长度,利于活性氧物种的迁移,同时在一定程度上阻止活性Mn位点的氯化;另一方面,Co的适量掺入显著增强了材料表面的L酸和B酸位点,进一步促进了污染物的吸附活化与氯物种的及时脱附. 在催化降解HCBD过程中,污染物首先吸附在L酸位点,随后被活性氧物种攻击,生成丙酮、甲醛、二氯乙烷以及含有酯键和醚键等含氧官能团的中间产物,进一步在酸性位点和活性氧的作用下被深度降解.
-
本文基于锰基莫来石SmMn2O5(AB2O5)进行B位调控,研究在B位主要元素为Mn的前提下,Co元素不同掺杂占比调控对HCBD降解活性的影响,分析了各催化体系的反应动力学,研究了B位Co掺杂引入后催化剂的晶相组成、微观形貌、表面化学性质等结构特征,阐明了调控机理,结合界面反应过程的实时监测,揭示了B位调控钐锰莫来石催化剂对HCBD的催化降解机制. 具体结论如下:
1)与SmMn2O5相比,Co 的引入在一定程度上提高了Sm(Mn1-xCox)2O5(0<x≤0.5)系列催化剂对HCBD 的降解效率和反应速率. 其中Sm(Mn0.8Co0.2)2O5表现出优异的降解活性,60 min时对HCBD的降解效率约66%,同时反应符合准一级动力学,反应速率常数为0.019 min−1.
2) B位Co的引入能改变Mn的配位环境,具体表现为,Co掺杂占比较低时,Co取代B位部分Mn,产生了高活性的Co2+—O—Mn4+位点;随着掺杂占比的增加(0.2—0.3),形成了新的物相SmMnO3;当Co对Mn取代接近饱和时,额外形成SmCoO3(0.4—0.5).
3)适量Co的引入产生的Co2+—O—Mn4+位点削弱了金属氧键,导致原本的Mn—O键拉长,促进活性氧的移动性,有利于氧的迁移和活性氧物种的生成,在一定程度上阻止活性Mn位点被氯化失活,同时显著增多材料表面的L酸和B酸位点. 在活性氧物种和酸性位点作用下,HCBD首先生成丙酮、甲醛、二氯乙烷以及含有酯键和醚键等含氧官能团的中间产物,随后被进一步被降解.
B位Co掺杂调控SmMn2O5催化剂对六氯丁二烯的热催化降解机制研究
Research on the mechanism of thermal catalytic degradation of hexachlorobutadiene over B-site Co-doping SmMn2O5 catalysts
-
摘要: 开展六氯丁二烯(Hexachlorobutadiene,HCBD)的热催化降解研究,对于有效推进氯代持久性有机污染物控制技术应用的发展具有重要意义. 本研究系统探究了B位Co不同掺杂占比对SmMn2O5(AB2O5)催化降解活性的调控. 在300 ℃下,Co 的引入能在一定程度上提高Sm(Mn1-xCox)2O5 (0<x≤0.5)对HCBD的降解活性,其中Sm(Mn0.8Co0.2)2O5表现较为优异,在反应60 min时HCBD的降解效率和准一级反应动力学常数分别为66% 和0.019 min−1. 通过对催化剂结构特征的综合表征,发现B位适量Co的引入取代了部分Mn3+,形成了Co2+—O—Mn4+活性位点,改变了SmMn2O5中Mn的配位环境,增多了材料表面活性氧物种含量并提高了其迁移性,同时显著增强了材料表面的Lewis酸和Brnsted酸位点. 在活性氧物种和酸位的作用下,HCBD被降解为丙酮、甲醛、二氯乙烷及含酯键和醚键等含氧官能团的中间产物,随后被进一步降解. 本研究为氯代有机污染物的削减控制提供了重要的技术支撑.Abstract: The elucidation of thermo-catalytic degradation routes for hexachlorobutadiene (HCBD), a chlorinated persistent organic pollutant, is pivotal for the advancement of pollution control technologies. This research conducts an exhaustive analysis of the Co-doped catalytic activity regulatory mechanisms in SmMn2O5 (AB2O5) by manipulating the doping ratio of Co at the B-site. At a temperature threshold of 300°C, the strategic infusion of Co was observed to elevate the HCBD degradation efficacy of Sm(Mn1-xCox)2O5 (0<x≤0.5), with the optimal Sm(Mn0.8Co0.2)2O5 demonstrating an enhanced performance, culminating in a degradation efficiency of 66% and a pseudo-first-order reaction rate constant of 0.019 min−1 after a reaction duration of 60 min. A thorough structural analysis of the catalyst illuminated that the moderate introduction of Co to the B-site ousted some Mn3+ ions, engendering Co2+—O—Mn4+ sites of activity, consequently modifying the coordination context of Mn within SmMn2O5. This action proliferated the surficial active oxygen species and improved their mobility, significantly augmenting the Lewis acid and Brønsted acid sites evident on the catalyst's surface. Under the synergistic impact of these active oxygen species and associated acidic sites, HCBD is decomposed to intermediate compounds, inclusive of acetone, formaldehyde, dichloroethane, and entities bearing ester and ether bonds, promoting further degradation. This study provides important technical support for the reduction and control of chlorinated organic pollutants.
-

-
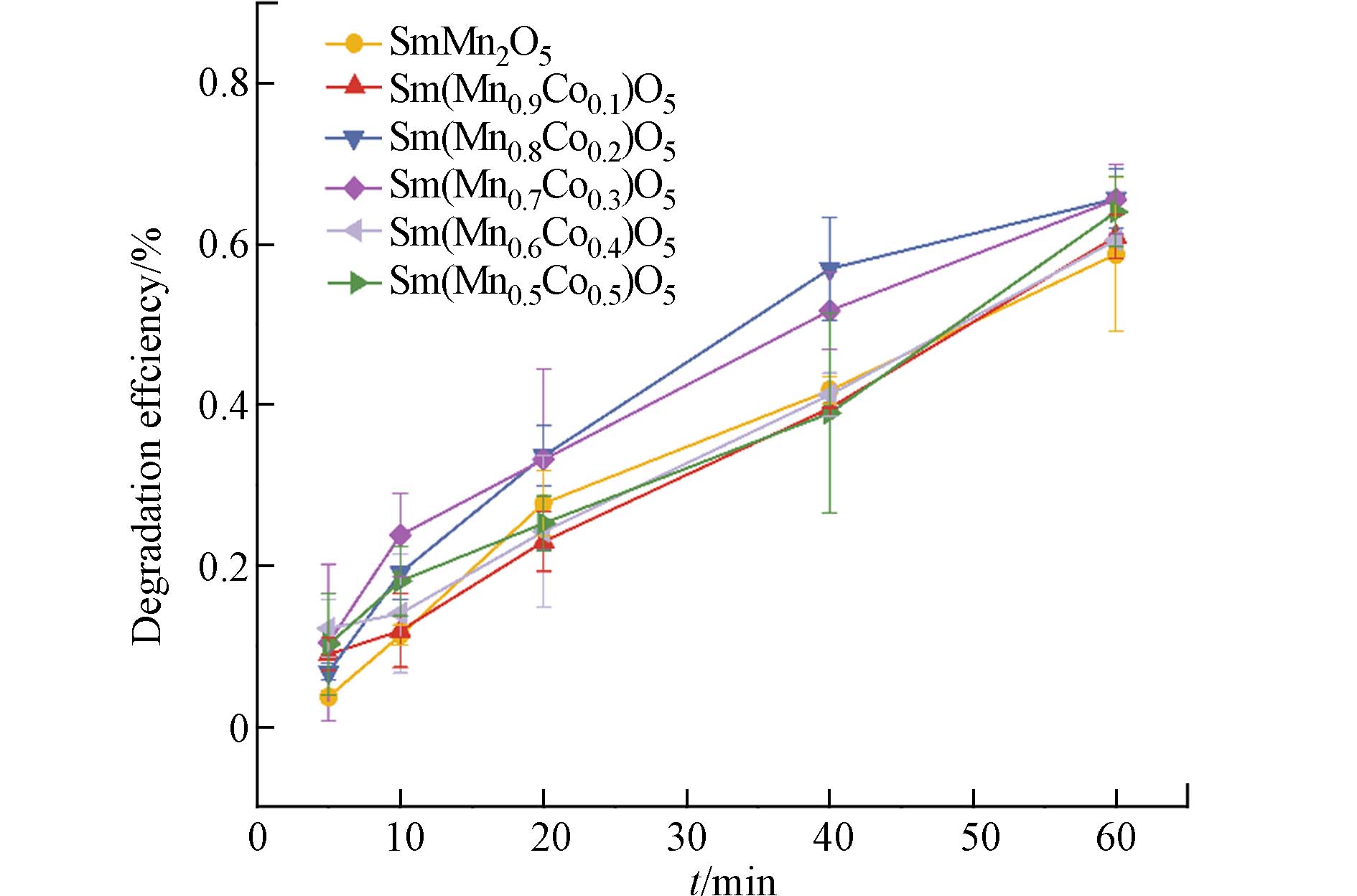
图 1 Sm(Mn1-xCox)2O5 (0≤x≤0.5)系列催化剂对HCBD降解活性评价
Figure 1. Evaluation of the degradation activity of Sm(Mn1-xCox)2O5 (0≤x≤0.5) series catalysts on HCBD
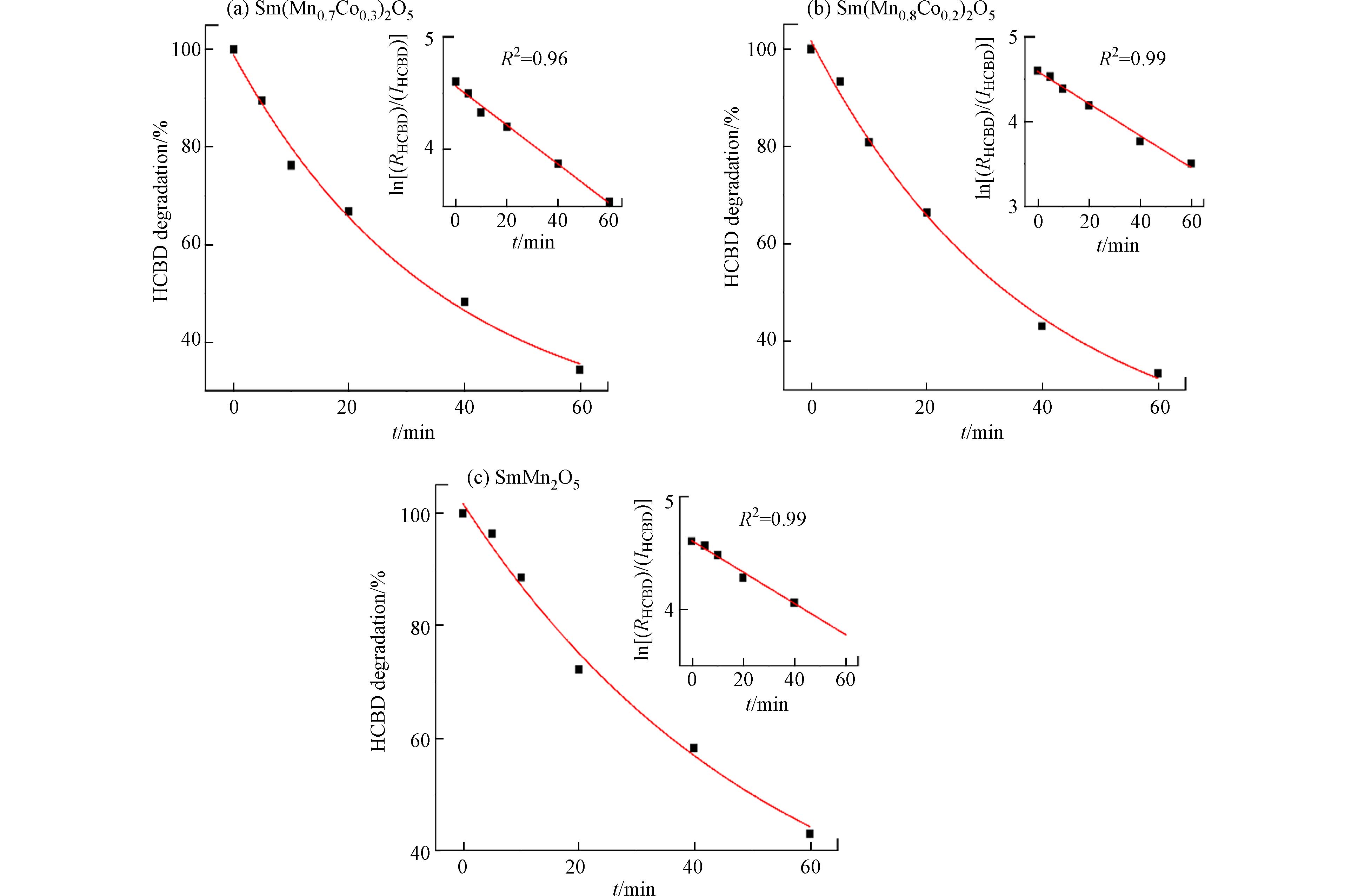
图 2 在300°C下(a) Sm(Mn0.8Co0.2)2O5、(b) Sm(Mn0.7Co0.3)2O5和(c)SmMn2O5催化体系中HCBD降解率反应时间的变化曲线及拟合的准一级动力学直线图
Figure 2. The change curve of HCBD degradation rate reaction time in (a) Sm(Mn0.8Co0.2)2O5、(b) Sm(Mn0.7Co0.3)2O5 and (c)SmMn2O5 catalytic systems at 300 °C and the fitted quasi-first-order kinetic linear graph
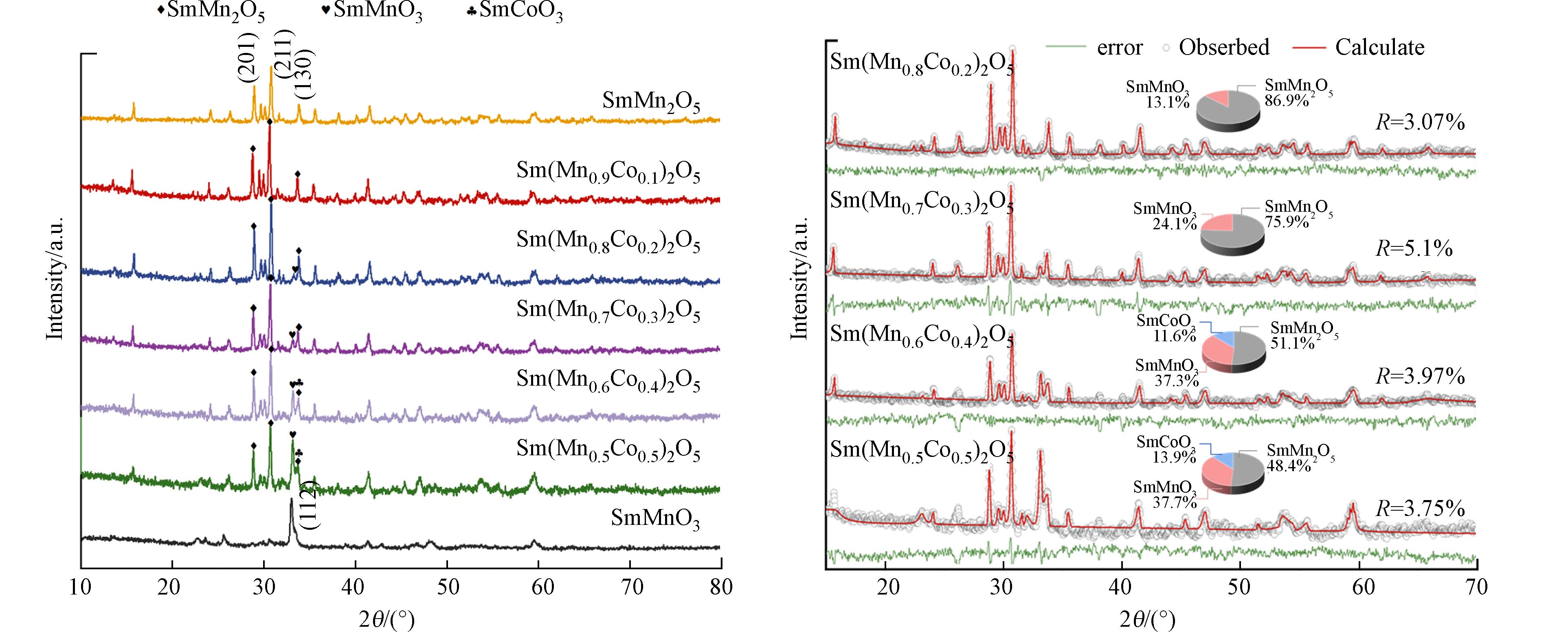
图 3 Sm(Mn1-xCox)2O5(0≤x≤0.5)系列催化XRD 图谱(a)和XRD精修图谱(b)
Figure 3. XRD pattern of Sm(Mn1-xCox)2O5(0≤x≤0.5)series catalysts
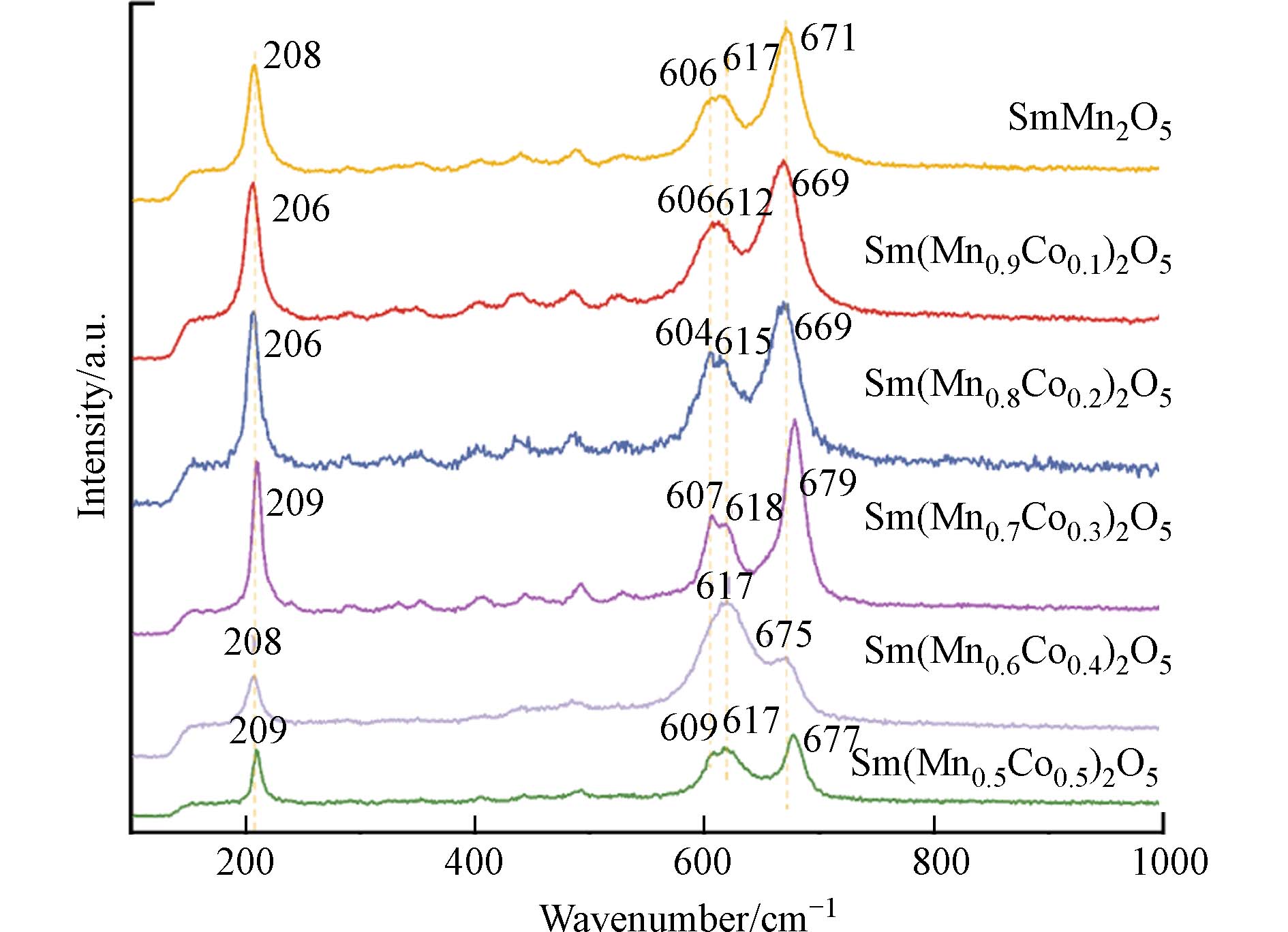
图 4 Sm(Mn1-xCox)2O5(0≤x≤0.5) 系列部分催化剂 Raman 图谱
Figure 4. Raman spectra of Sm(Mn1-xCox)2O5(0≤x≤0.5) series catalysts.
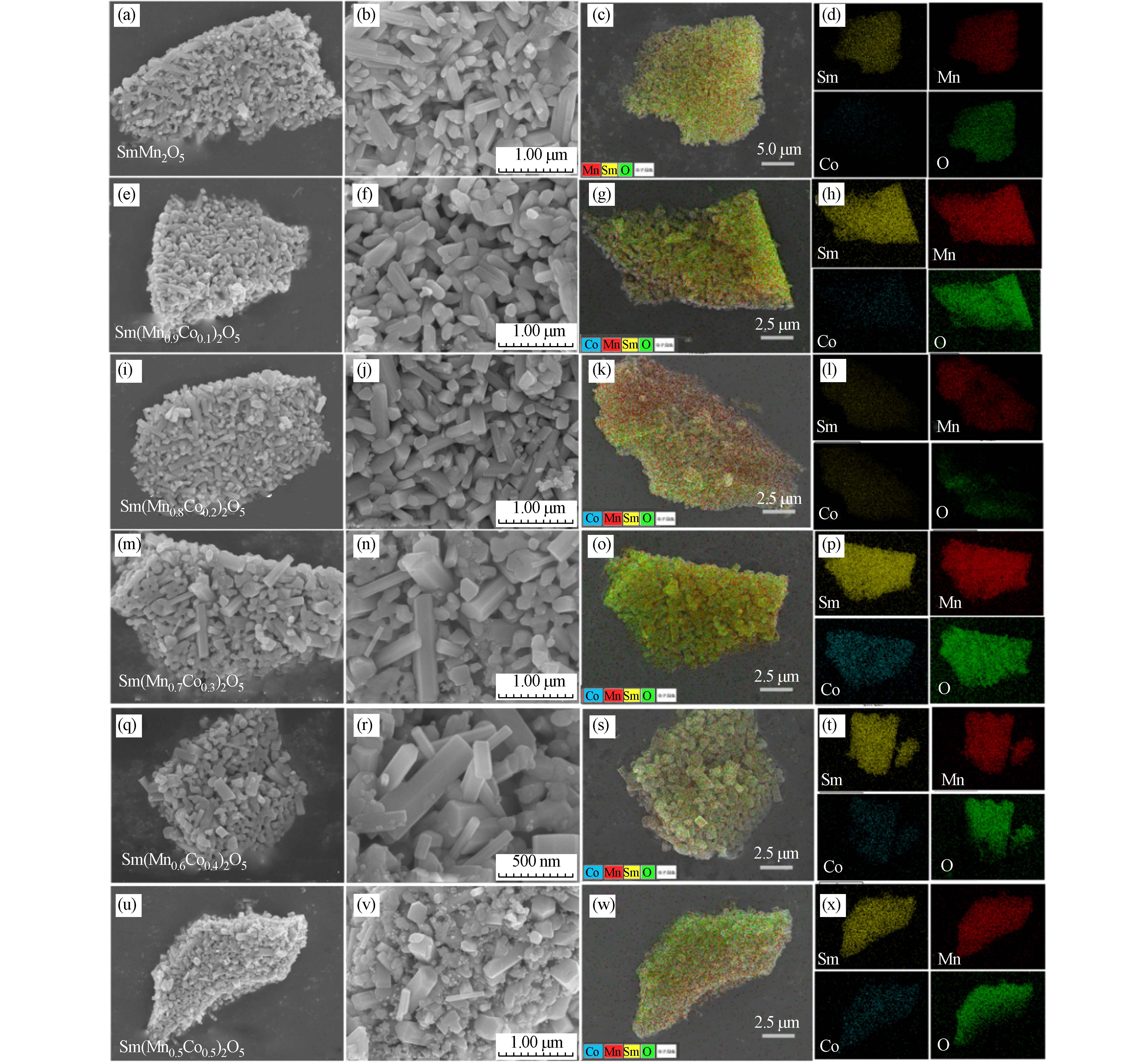
图 5 Sm(Mn1-xCox)2O5(0≤x≤0.5)系列催化剂SEM 以及单元素 EDS mapping 图谱 :SmMn2O5 (a,b,c,d); Sm(Mn0.9Co0.1)2O5(e,f,g,h) ; Sm(Mn0.8Co0.2)2O5(i,j,k,l) ; Sm(Mn0.7Co0.3)2O5(m,n,o,p); Sm(Mn0.6Co0.4)2O5(q,r,s,t);Sm(Mn0.5Co0.5)2O5(u,v,w,x).
Figure 5. SEM and EDS mapping of Sm(Mn1-xCox)2O5(0≤x≤0.5)series catalysts;SmMn2O5 (a,b,c,d); Sm(Mn0.9Co0.1)2O5(e,f,g,h) ; Sm(Mn0.8Co0.2)2O5(i,j,k,l) ; Sm(Mn0.7Co0.3)2O5(m,n,o,p); Sm(Mn0.6Co0.4)2O5(q,r,s,t);Sm(Mn0.5Co0.5)2O5(u,v,w,x).
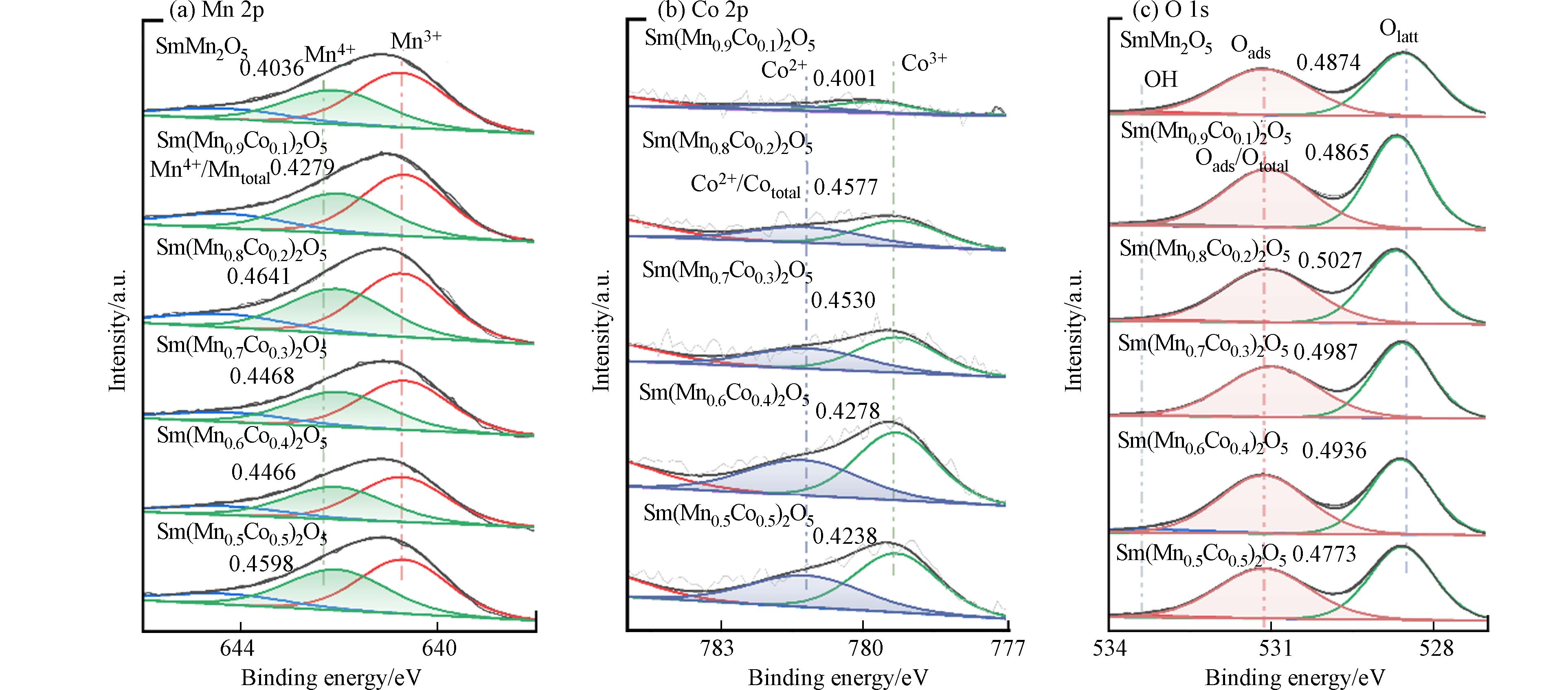
图 6 Sm(Mn1-xCox)2O5(0≤x≤0.5)系列部分催化剂Mn 2p (a) , Co 2p (b) 和O1s (c)XPS 图谱
Figure 6. XPS spectra of partial catalysts Mn 2p (a), Co 2p (b) and O1s(c)in Sm(Mn1-xCox)2O5(0≤x≤0.5)series
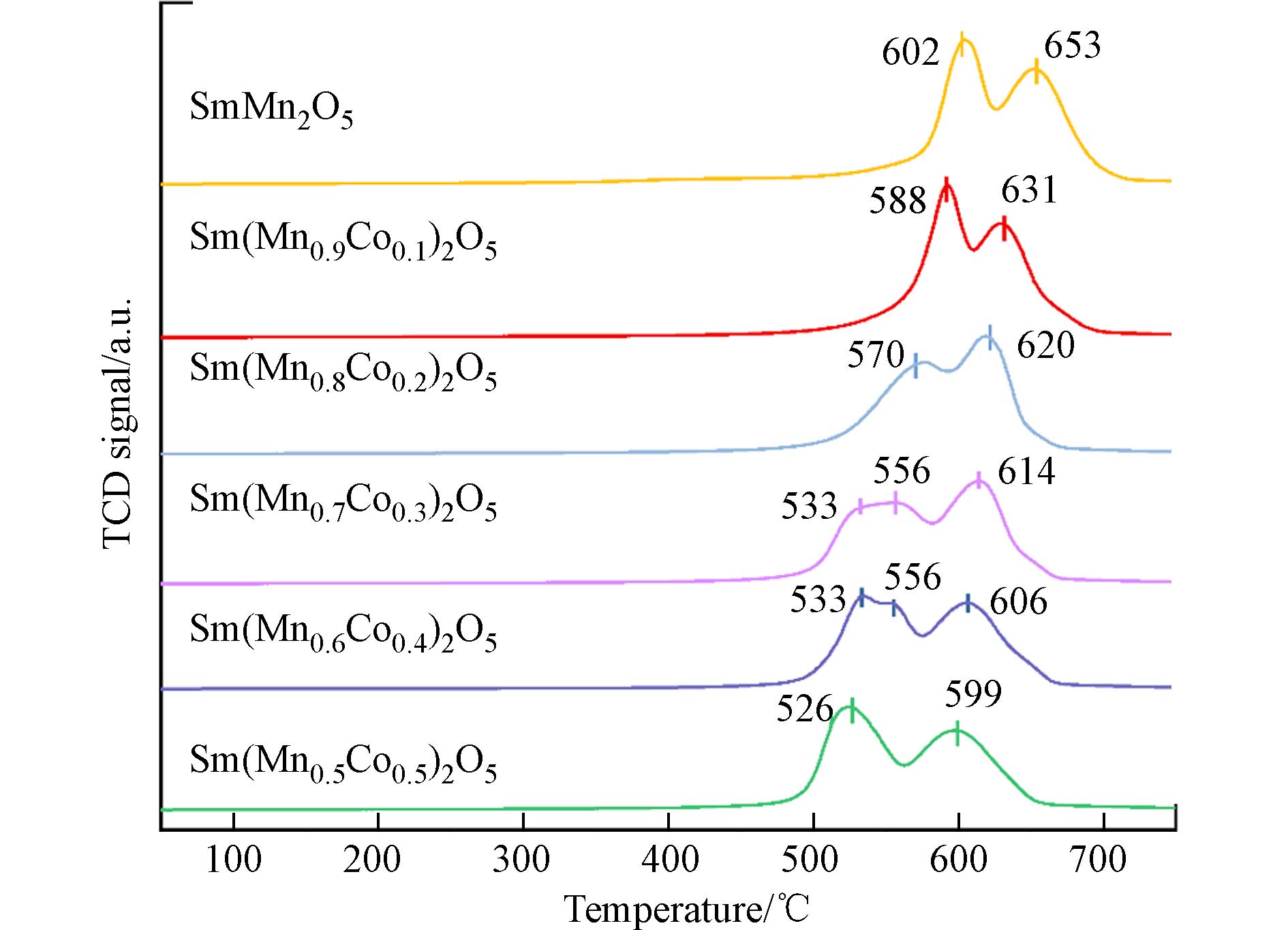
图 7 Sm(Mn1-xCox)2O5 (0≤x≤0.5)系列催化剂H2—TPR
Figure 7. H2—TPR of Sm(Mn1-xCox)2O5 (0≤x≤0.5)
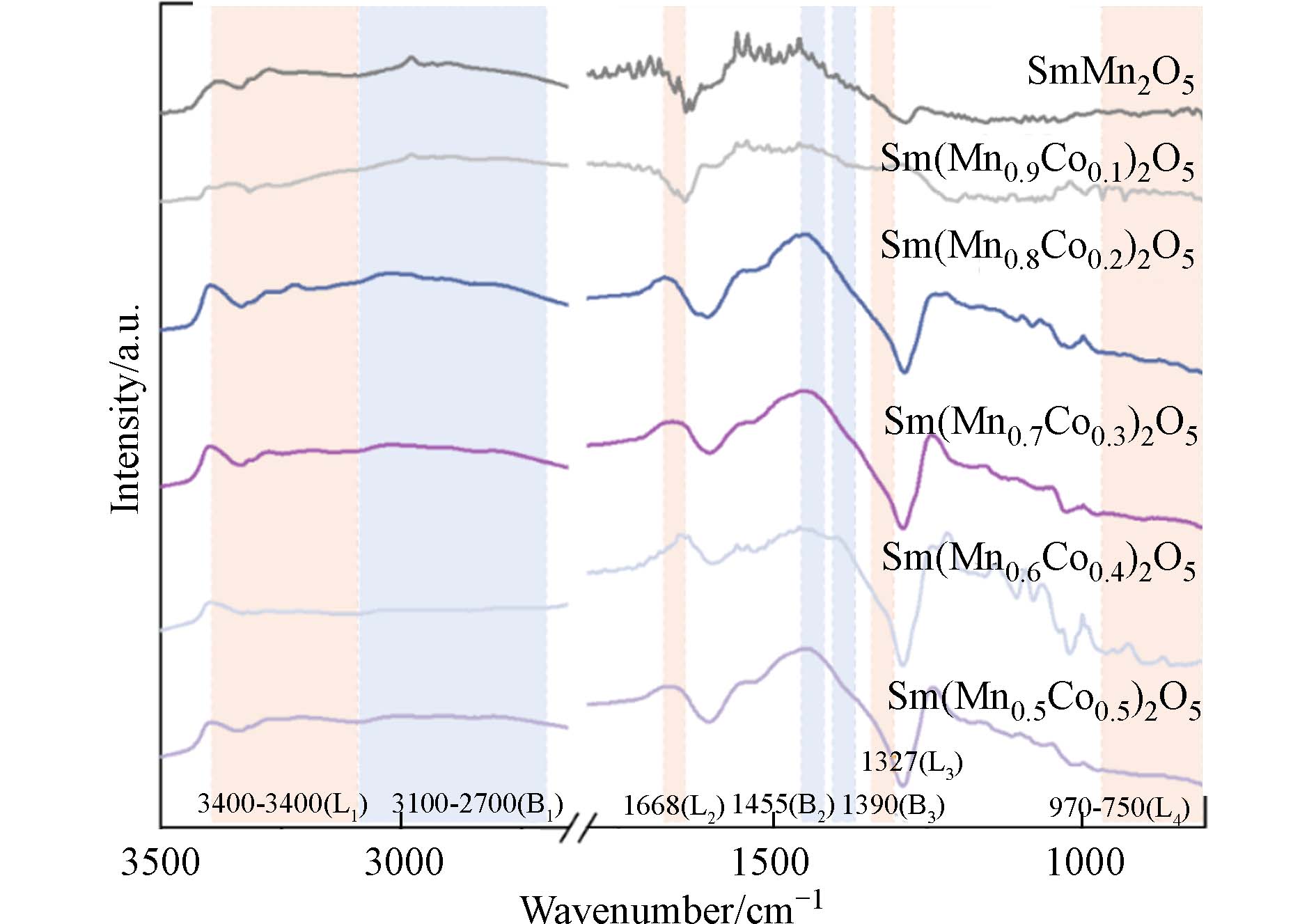
图 8 Sm(Mn1-xCox)2O(0≤x≤0.5)系列催化剂NH3—FTIR图
Figure 8. NH3-FTIR spectra of Sm(Mn1-xCox)2O5 (0≤x≤0.5) catalysts
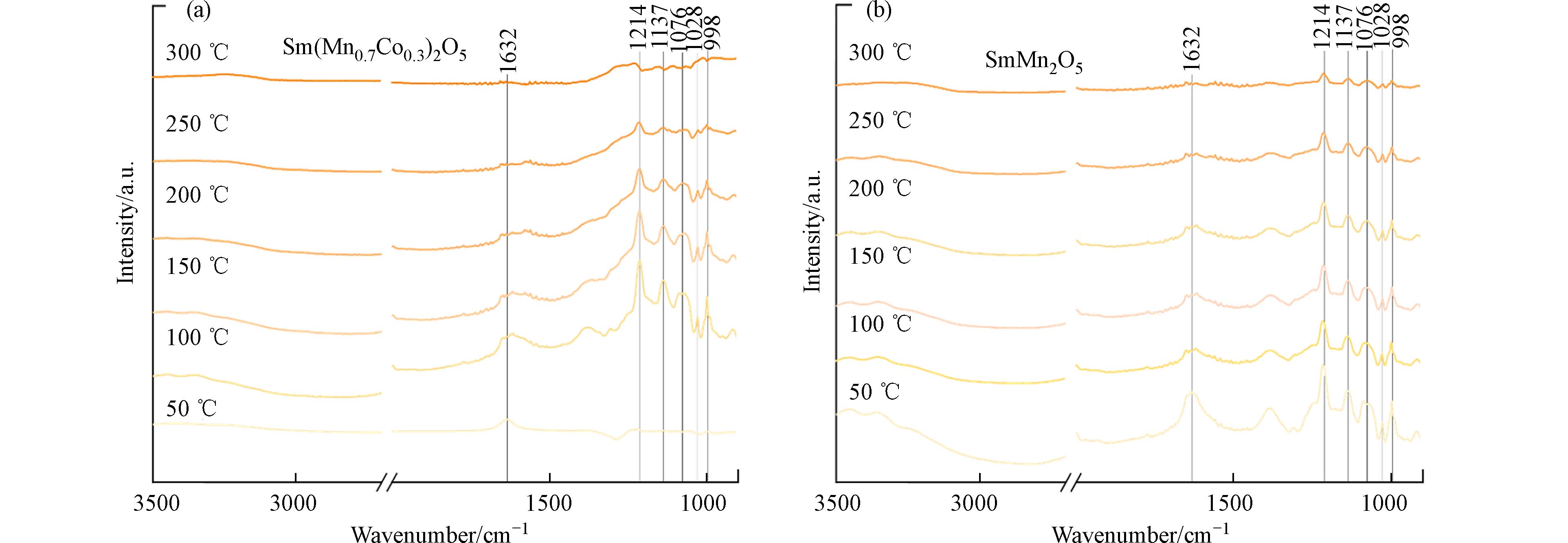
图 9 (a)Sm(Mn0.7Co0.3)O5和(b)SmMn2O5不同温度的原位红外光谱图
Figure 9. In situ DRIFTS of (a) Sm(Mn0.7Co0.3)O5 and (b) SmMn2O5 i at different temperatures
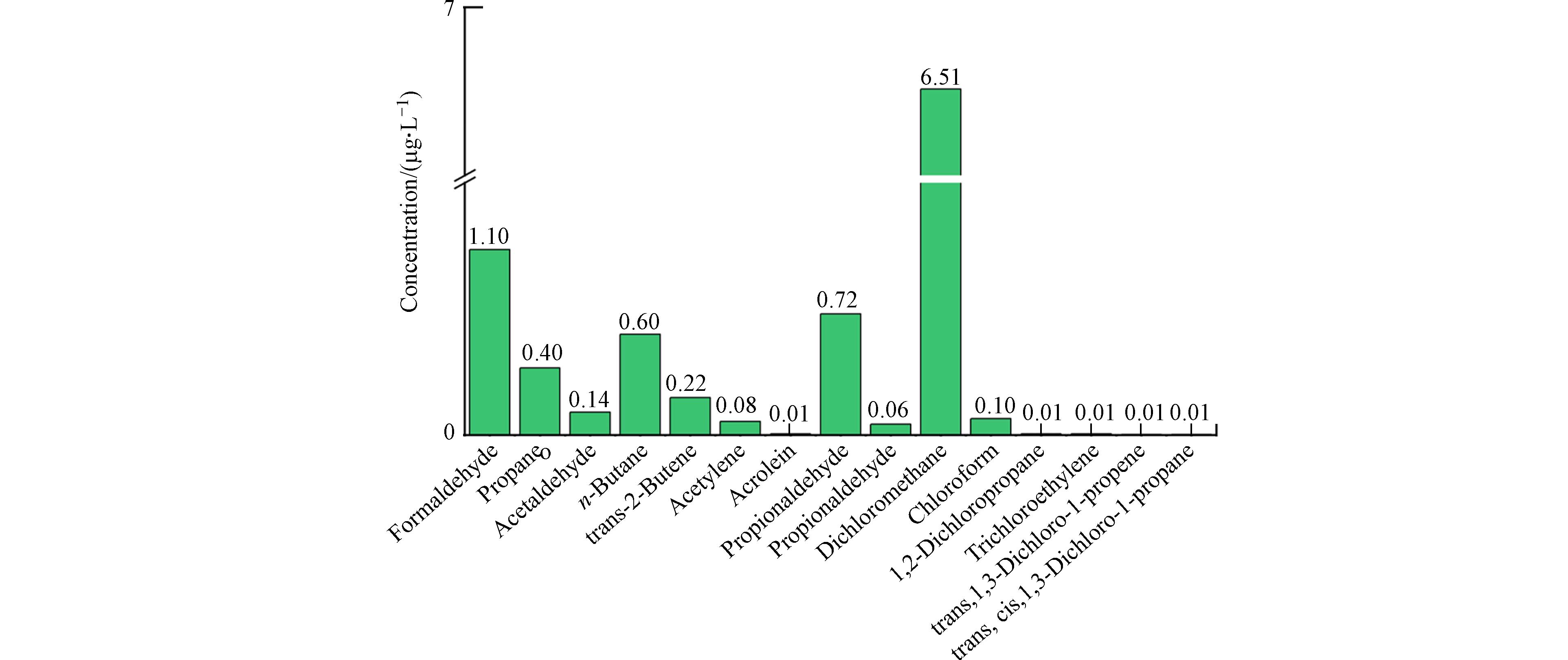
图 10 300 ℃条件下Sm(Mn0.8Co0.2)2O5降解HCBD的气相产物定量分析结果
Figure 10. Quantitative analysis of gaseous products of HCBD degraded by Sm(Mn0.8Co0.2)2O5 at 300 ℃
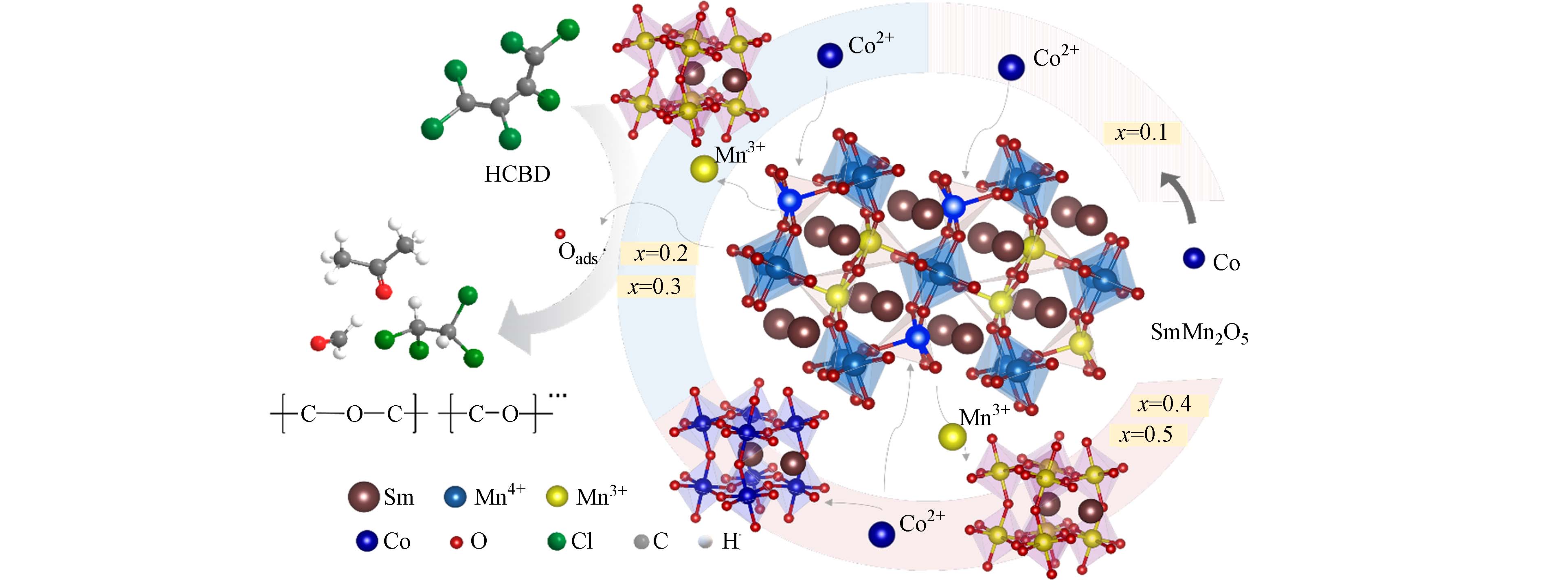
图 11 锰基莫来石SmMn2O5(AB2O5)中B位Co引入Sm(Mn1-xCox)2O5(0≤x≤0.5)系列材料对HCBD的降解机理图
Figure 11. The degradation mechanism diagram of HCBD by incorporating Co into the B-site of mangano-mullite SmMn2O5 (AB2O5) to form the Sm(Mn1-xCox)2O5(0≤x≤0.5) series catalysis
表 1 Sm(Mn1-xCox)2O5(0≤x≤0.5)催化降解HCBD的反应速率常数
Table 1. Rate constant of catalytic degradation of HCBD by Sm(Mn1-xCox)2O5(0≤x≤0.5) series catalysies
不同掺杂配比材料
Materials with different doping ratios反应速率常数kobs / min−1
Reaction rate constantR2 SmMn2O5 0.0142 0.99 Sm(Mn0.9Co0.1)2O5 0.0149 0.98 Sm(Mn0.8Co0.2)2O5 0.019 0.99 Sm(Mn0.7Co0.3)2O5 0.0173 0.99 Sm(Mn0.6Co0.4)2O5 0.0146 0.99 Sm(Mn0.5Co0.5)2O5 0.0156 0.96  下载: 导出CSV
下载: 导出CSV
表 2 Sm(Mn1-xCox)2O(0≤x≤0.5)系列催化剂比表面积表征结果
Table 2. BET results of Sm(Mn1-xCox)2O5 (0≤x≤0.5) series catalysts
不同掺杂配比材料
Materials with different doping ratios比表面积/(m2·g−1)
SBET孔体积/(cm3·g−1)
Pore volume平均孔径/nm
Mean pore diameterSmMn2O5 33.09 0.02 16.36 Sm(Mn0.9Co0.1)2O5 11.16 0.0048 6.7643 Sm(Mn0.8Co0.2)2O5 14.96 0.0039 4.7361 Sm(Mn0.7Co0.3)2O5 15.99 0.0037 4.424+ Sm(Mn0.6Co0.4)2O5 6.87 0.0022 3.1167 Sm(Mn0.5Co0.5)2O5 2.52 0.0197 5.4642
下载: 导出CSV
表 3 部分材料反应后表面氯元素含量
Table 3. Chlorine content on the surface of some materials after reaction
XPS半定量/% Sm(Mn0.8Co0.2)2O5 7.03 Sm(Mn0.6Co0.4)2O5 7.46 Sm(Mn0.5Co0.5)2O5 7.67
下载: 导出CSV
-
[1] BALMER J E, HUNG H, VORKAMP K, et al. Hexachlorobutadiene (HCBD) contamination in the Arctic environment: A review[J]. Emerging Contaminants, 2019, 5: 116-122. doi: 10.1016/j.emcon.2019.03.002 [2] 金慧娟, 杨毅, 李秀颖, 等. 六氯-1, 3-丁二烯的微生物降解研究进展[J]. 微生物学通报, 2020, 47(10): 3407-3418. JIN H J, YANG Y, LI X Y, et al. Progress in microbial degradation of hexachlorobutadiene[J]. Microbiology China, 2020, 47(10): 3407-3418 (in Chinese).
[3] WANG M X, YANG L L, LIU X Y, et al. Hexachlorobutadiene emissions from typical chemical plants[J]. Frontiers of Environmental Science & Engineering, 2020, 15(4): 60. [4] WANG L, BIE P J, ZHANG J B. Estimates of unintentional production and emission of hexachlorobutadiene from 1992 to 2016 in China[J]. Environmental Pollution, 2018, 238: 204-212. doi: 10.1016/j.envpol.2018.03.028 [5] BOOKER R. Microbial reductive dechlorination of hexachloro-1, 3-butadiene in a methanogenic enrichment culture[J]. Water Research, 2000, 34(18): 4437-4445. doi: 10.1016/S0043-1354(00)00214-1 [6] LI M T, HAO L L, SHENG L X, et al. Identification and degradation characterization of hexachlorobutadiene degrading strain Serratia marcescens HL1[J]. Bioresource Technology, 2008, 99(15): 6878-6884. doi: 10.1016/j.biortech.2008.01.048 [7] BOSMA T N, COTTAAR F H, POSTHUMUS M A, et al. Comparison of reductive dechlorination of hexachloro-1, 3-butadiene in Rhine sediment and model systems with hydroxocobalamin[J]. Environmental Science & Technology, 1994, 28(6): 1124-1128. [8] 王庆良. 1, 3-丁二烯及六氯丁二烯的光化学反应研究[D]. 成都: 成都理工大学, 2020. WANG Q L. Photochemical reactions of 1, 3-butadiene and hexachlorobutadiene[D]. Chengdu: Chengdu University of Technology, 2020 (in Chinese).
[9] 赵晨妍, 孙宇翔, 杨莉莉, 等. 六氯丁二烯的排放源及环境污染特征[J]. 化学进展, 2023, 35(7): 1040-1052. doi: 10.7536/PC221126 ZHAO C Y, SUN Y X, YANG L L, et al. Source and environmental characteristics of hexachlorobutadiene[J]. Progress in Chemistry, 2023, 35(7): 1040-1052 (in Chinese). doi: 10.7536/PC221126
[10] 生态环境部, 中国政府网, 重点管控新污染物清单(2023年版)[EB] 2024-02-19]. Ministry of Ecology and Environment, List of New Pollutants under Key Control of the Chinese Government Network (2023 version). [EB][2024-02-19].
[11] 陶誉铭. 改性莫来石型催化剂制备及对六氯丁二烯降解性能研究[D]. 长春: 长春理工大学, 2021. TAO Y M. Preparation of modified mullite catalyst and degradation of hexachlorobutadiene[D]. Changchun: Changchun University of Science and Technology, 2021 (in Chinese).
[12] YEE L H, AAGAARD V, JOHNSTONE A, et al. Development of a treatment solution for reductive dechlorination of hexachloro-1, 3-butadiene in vadose zone soil[J]. Biodegradation, 2010, 21(6): 947-956. doi: 10.1007/s10532-010-9354-z [13] KONG Q Q, WANG Y, YANG X. A review on hexachloro-1, 3-butadiene (HCBD): Sources, occurrence, toxicity and transformation[J]. Bulletin of Environmental Contamination and Toxicology, 2020, 104(1): 1-7. doi: 10.1007/s00128-019-02744-5 [14] LIU Y, HU H L, ZHENG J M, et al. Interfacial engineering enables surface lattice oxygen activation of SmMn2O5 for catalytic propane combustion[J]. Applied Catalysis B: Environmental, 2023, 330: 122649. doi: 10.1016/j.apcatb.2023.122649 [15] WANG W C, McCOOL G, KAPUR N, et al. Mixed-phase oxide catalyst based on Mn-mullite (Sm, Gd)Mn2O5 for NO oxidation in diesel exhaust[J]. Science, 2012, 337(6096): 832-835. doi: 10.1126/science.1225091 [16] WANG F L, WANG P L, LAN T W, et al. Ultralow-temperature NOx reduction over SmMn2O5 mullite catalysts via modulating the superficial dual-functional active sites[J]. ACS Catalysis, 2022, 12(13): 7622-7632. doi: 10.1021/acscatal.2c01897 [17] SHEN Y J, DENG J, HAN L P, et al. Low-temperature combustion of toluene over Cu-doped SmMn2O5 mullite catalysts via creating highly active Cu2+-O-Mn4+ sites[J]. Environmental Science & Technology, 2022, 56(14): 10433-10441. [18] GOLOVENCHITS E, SANINA V. Magnetic and magnetoelectric dynamics in RMn2O5 (R = Gd and Eu)[M]. Magnetoelectric Interaction Phenomena in Crystals. Dordrecht: Springer Netherlands, 2004: 139-150. [19] GARCÍA-FLORES A F, GRANADO E, MARTINHO H, et al. Anomalous phonon shifts in the paramagnetic phase of multiferroic RMn2O5(R=Bi, Eu, Dy): Possible manifestations of unconventional magnetic correlations[J]. Physical Review B, 2006, 73(10): 104411. doi: 10.1103/PhysRevB.73.104411 [20] LI W L, MAO H, JIN B F, et al. High-surface-area SmMn2O5 nanosheets with crystal orientation for propane combustion: A facile microwave-assisted hydrothermal method[J]. Fuel, 2021, 306: 121685. doi: 10.1016/j.fuel.2021.121685 [21] THAMPY S, ASHBURN N, DILLON S, et al. Critical role of mullite-type oxides surface chemistry on catalytic NO oxidation performance[J]. The Journal of Physical Chemistry C, 2019, 123(9): 5385-5393. doi: 10.1021/acs.jpcc.8b10670 [22] CHEN X, YANG J Q, LIU Z, et al. Origin of ammonia selective oxidation activity of SmMn2O5 mullite: A first-principles-based microkinetic study[J]. ACS Applied Materials & Interfaces, 2023, 15(1): 736-750. [23] FENG Z J, WANG J Q, LIU X, et al. Promotional role of La addition in the NO oxidation performance of a SmMn2O5 mullite catalyst[J]. Catalysis Science & Technology, 2016, 6(14): 5580-5589. [24] YANG J Q, ZHANG J, LIU X, et al. Origin of the superior activity of surface doped SmMn2O5 mullites for NO oxidation: A first-principles based microkinetic study[J]. Journal of Catalysis, 2018, 359: 122-129. doi: 10.1016/j.jcat.2018.01.002 [25] YANG Q L, LI Q, WANG X Y, et al. Synergistic effects of a CeO2/SmMn2O5-H diesel oxidation catalyst induced by acid-selective dissolution drive the catalytic oxidation reaction[J]. ACS Applied Materials & Interfaces, 2022, 14(2): 2860-2870. [26] MANTILLA J, MORALES M, VENCESLAU W, et al. Field-driven spin reorientation in SmMnO3 polycrystalline powders[J]. Journal of Alloys and Compounds, 2020, 845: 156327. doi: 10.1016/j.jallcom.2020.156327 [27] ZHU G Q, LIU P, HOJAMBERDIEV M, et al. Synthesis RMn2O5 (R = Gd and Sm) nano- and microstructures by a simple hydrothermal method[J]. Materials Chemistry and Physics, 2009, 118(2-3): 467-472. doi: 10.1016/j.matchemphys.2009.08.019 [28] YANG Q L, WANG X Y, LI X B, et al. Surface tailoring on SrMnO3@SmMn2O5 for boosting the performance in diesel oxidation catalyst[J]. Materials Chemistry and Physics, 2009, 118(2-3): 467-472. doi: 10.1016/j.matchemphys.2009.08.019 [29] MORALES M R, BARBERO B P, CADÚS L E. Total oxidation of ethanol and propane over Mn-Cu mixed oxide catalysts[J]. Applied Catalysis B, Environmental, 2006, 67(3): 229-236. [30] MACHOCKI A, IOANNIDES T, STASINSKA B, et al. Manganese–lanthanum oxides modified with silver for the catalytic combustion of methane[J]. Journal of Catalysis, 2004, 227(2): 282-296. doi: 10.1016/j.jcat.2004.07.022 [31] WU H, PANTALEO G, Di CARLO G, et al. Co3O4 particles grown over nanocrystalline CeO2: Influence of precipitation agents and calcination temperature on the catalytic activity for methane oxidation[J]. Catalysis Science & Technology, 2015, 5(3): 1888-1901. [32] PU Z Y, LIU Y, ZHOU H, et al. Catalytic combustion of lean methane at low temperature over ZrO2-modified Co3O4 catalysts[J]. Applied Surface Science, 2017, 422: 85-93. doi: 10.1016/j.apsusc.2017.05.231 [33] 冯子健. 莫来石型氧化物SmMn2O5在柴油车尾气处理和甲烷燃烧中的催化性能研究[D]. 武汉: 华中科技大学, 2018. FENG Z J. Catalytic performance of SmMn2O5 mullite for diesel exhaust purification and methane combustion[D]. Wuhan: Huazhong University of Science and Technology, 2018 (in Chinese).
[34] WU H F, ZHANG W J, LIU Y W, et al. One-step control of Brønsted acid sites and oxygen vacancies in Mn-based perovskite for boosting catalytic oxidation of chlorobenzene[J]. Journal of Environmental Chemical Engineering, 2023, 11(3): 110210. doi: 10.1016/j.jece.2023.110210 [35] LIU L Z, ZHOU B, LIU Y W, et al. In-situ regulation of acid sites on Mn-based perovskite@mullite composite for promoting catalytic oxidation of chlorobenzene[J]. Journal of Colloid and Interface Science, 2022, 606: 1866-187. doi: 10.1016/j.jcis.2021.08.145 [36] GHIASSEE M, REZAEI M, MESHKANI F, et al. Preparation and optimization of the MnCo2O4 powders for low temperature CO oxidation using the Taguchi method of experimental design[J]. Research on Chemical Intermediates, 2019, 45(9): 4501-4515. doi: 10.1007/s11164-019-03845-w [37] SI W Z, WANG Y, ZHAO S, et al. A facile method for in situ preparation of the MnO2/LaMnO3 catalyst for the removal of toluene[J]. Environmental Science & Technology, 2016, 50(8): 4572-4578. [38] LIU R Y, ZHOU B, LIU L Z, et al. Enhanced catalytic oxidation of VOCs over porous Mn-based mullite synthesized by in situ dismutation[J]. Journal of Colloid and Interface Science, 2021, 585: 302-311. doi: 10.1016/j.jcis.2020.11.096 [39] YAFAROVA LILIYA V, MAMONTOV GRIGORY V, CHISLOVA IRINA V, et al. The effect of transition metal substitution in the perovskite-type oxides on the physicochemical properties and the catalytic performance in diesel soot oxidation[J]. Catalysts, 2021, 11(10): 1256. doi: 10.3390/catal11101256 [40] PANCHENKO Y N, GREKINA O E, MOCHALOV V I, et al. Vibrational spectra and conformational analysis of five chlorosubstituted buta-1, 3-dienes[J]. Journal of Molecular Structure, 1978, 49(1): 17-27. doi: 10.1016/0022-2860(78)87003-3 [41] YANG P, ZUO S F, ZHOU R X. Synergistic catalytic effect of (Ce, Cr)xO2 and HZSM-5 for elimination of chlorinated organic pollutants[J]. Chemical Engineering Journal, 2017, 323: 160-170. doi: 10.1016/j.cej.2017.04.002 [42] WAN X, WANG L, GAO S, et al. Low-temperature removal of aromatics pollutants via surface labile oxygen over Mn-based mullite catalyst SmMn2O5[J]. Chemical Engineering Journal, 2021, 410: 128305. doi: 10.1016/j.cej.2020.128305 [43] WANG J, WANG X, LIU X L, et al. Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts: The effects of chlorine substituents[J]. Catalysis Today, 2015, 241: 92-99. doi: 10.1016/j.cattod.2014.04.002 [44] GUNDERSEN G. Molecular structure of gaseous hexachlorobutadiene[J]. Journal of the American Chemical Society, 1975, 97(22): 6342-6346. doi: 10.1021/ja00855a009 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 124
- HTML全文浏览数: 124
- PDF下载数: 0
- 施引文献: 0
