

-
氢氯氟烃 (HCFCs)曾被广泛地应用到空调、制冷、泡沫、气溶胶推进剂和阻燃剂等多个领域[1-2]. 因HCFCs会破坏臭氧层,被《蒙特利尔议定书》列为受控物质[3],所以一些化合物常被用来替代HCFCs[4],其中以氢氟烃类(HFCs)和氢氟烯烃类(HFOs)为代表化合物. 在过去几年中,这些替代物的排放量呈现快速增长的势头,并不断释放到大气中[4-5]. 为评估这些替代物是否为HCFCs的理想替代,在这些化合物进入大气对流层之前,需要充分了解其大气氧化机制的完整信息,尤其是大气持久性.
大气中的·OH具有强氧化性和低选择性,是很多污染物氧化降解的关键物种[6],因此污染物与·OH反应的二级反应速率常数(kOH, cm3·molecule−1·s−1)是评价污染物大气持久性的重要参数. 传统的kOH实验测定方法耗时耗力,亟待发展新的方法获取HFCs和HFOs的kOH. 近年来,计算机软件、硬件的飞速提升和量子化学理论的不断发展,尤其是密度泛函理论(DFT),可直接从分子结构出发实现kOH从头计算. 采用适当的量子化学计算方法不仅速度快而且结果可以媲美实验值,因此有望在kOH的快速获取方面发挥重要作用,从而有助于评估污染物的大气持久性. 近年来,探究·OH引发气相污染物降解的反应机制和动力学的研究逐渐增多,包括丙酸甲酯[7]、多氯联苯[8]、农药[9]等.
此外,也不乏利用量子化学方法探究HFCs和HFOs大气转化机制的研究,这些研究涉及M06-2X/6-311++G(df,p)//6-31+G(df,p)、MP2/cc-pVDZ、M11/6-311++G(d,p)等多种DFT方法,和过渡态理论(TST)、正则变分过渡态理论、变分过渡态理论等多种kOH计算方法[10-14]. 然而,目前量子化学计算的研究均以探究大气转化机制为主要目标,而kOH作为大气转化过程中的一个参数很少有人系统研究其计算方法. 这些研究使用的量子化学方法通常仅针对单个HFCs或HFOs,对其它HFCs或HFOs是否适用仍未可知.
本研究考察了碳链长度、官能团位置等因素,选择3个HFCs:1,1,1,2-四氟乙烷(CF3CFH2)、1,1,1,3,3,3-六氟丙烷(CF3CH2CF3)、2H,3H-十氟戊烷(C2F5(CHF)2CF3)和2个HFOs:1,1-二氟乙烯(CF2=CH2)、1H,1H,2H-十七氟-1-葵烯(CF3(CF2)7CH2=CH2)作为模型化合物,以其kOH实测值为参考,筛选适用于计算HFCs和HFOs气相kOH值的量子化学方法. 将不同的方法计算所得kOH与实测值进行比较,发展关于HFCs和HFOs的高准确性和适用性的kOH计算方法.
-
为使筛选的计算方法能够广泛应用于HFCs和HFOs的气相kOH计算,本研究从文献已报道kOH的HFCs和HFOs中,根据碳链长度和官能团位置的不同,筛选出CF3CFH2[15]、CF3CH2CF3[15]、C2F5(CHF)2CF3[16]共3个HFCs和CF2=CH2[17]、CF3(CF2)7CH2=CH2[18]共2个HFOs作为模型化合物,用于筛选合适的计算方法,其结构如图1所示.
-
由于在大气中HFCs和HFOs存在多种构象,不同构象与·OH的反应性不同,因此采用波恩奥本海默分子动力学(BOMD)[19]模拟和量子化学计算相结合的方法来获得目标化合物的最稳定构象. BOMD模拟使用CP2K 8.2.0[20]软件包,NVT系综,利用Nose-Hoover控温方法将温度稳定在300 K,利用BLYPD3/DZVP-GTH方法计算5000步,步长为0.5 fs. 从模拟的动力学轨迹中选取多种能量较低的构象,然后使用M06-2X[21]/cc-pVDZ[22]计算方法对结构进行优化,最终选取能量最低的构象作为目标化合物的最稳定构象,用来考察它与·OH的反应,最低能量构象如图1所示. 量子化学计算在Gaussian 09[23]软件包中进行.
-
M06-2X泛函已证明能够很好地用于研究氢夺取反应[21, 24],因此,本研究中所有反应涉及的反应物、反应前络合物、过渡态、反应后络合物和产物均采用M06-2X/cc-pVDZ计算水平进行结构优化. 此外,通过M06-2X/cc-pVDZ方法计算内禀反应坐标验证过渡态的准确性.
对于单点能的计算,选用2种能够考察色散作用的泛函(M06-2X-D3和ωB97X-D)[25]与8种3-zeta基组(aug-cc-pVTZ、may-cc-pVTZ、jun-cc-pVTZ、jul-cc-pVTZ、def2-TZVP、def2-TZVPP、pcseg-2和MG3S)[22, 26]进行组合,总计16种计算方法.
-
本研究采用TST[27]计算模型化合物的每条反应通道的kOH :
式中,κ代表隧道效应修正系数; σ代表反应简并度;h代表普朗克常数(6.626 × 10−34 J·s);T代表温度(K);kB代表玻尔兹曼常数(1.381 × 10−23 J·K−1);R代表摩尔气体常数(8.314 J·mol−1·K−1);P0代表大气压强(105 Pa);∆G‡,0代表标准活化自由能(kJ·mol−1),为过渡态吉布斯自由能减去反应物吉布斯自由能;对于双分子反应∆n为1,模型化合物HFCs和HFOs的kOH为此物质所有反应通道kOH之和.
对于κ的值,选择两种方法:(Wigner隧道效应校正系数(κW)[28]和Skodje-Truhlar隧道效应校正系数(κS)[29-30])进行计算,进而筛选出最合适的计算κ的方法.
κW的计算公式如下:
式中,υi†是TS的虚频(cm−1).
κS的计算方法如下:
当α > β时:
当α = β时:
当α < β时:
其中,∆V为势垒高度(kJ·mol−1),∆E为过渡态减去反应物的能量(kJ·mol−1),当∆E > 0 kJ·mol−1时,∆V为0 kJ·mol−1;反之,∆V为产物减去反应物的能量.
-
理论上,·OH与HFCs、HFOs可以发生夺H原子或F原子的反应,还可以在不饱和键发生·OH加成反应. 前人在研究CF2=C(CH3)CF3、CF2=C(CH3)2、1H-七氟环戊烯与·OH的反应中发现·OH难以夺取F原子[10, 13],因此本研究仅考虑·OH夺取HFCs和HFOs上的H原子. 对于3个HFCs,其反应机理仅为夺取C原子上的H原子. 由于H原子的位置可能会影响·OH夺取能力,所以选择CF3CFH2、CF3CH2CF3以及C2F5(CHF)2CF3作为模型化合物. 考虑到CF3CFH2最低构象具有Cs对称性,与·OH反应仅计算1条氢夺取途径. 而C2F5(CHF)2CF3和CF3CH2CF3分子的最低构象不具备对称性,需考虑所有的氢夺取反应途径. 对于HFOs,·OH与其反应机理包括H夺取和·OH加成. 考虑到CF2=CH2和CF3(CF2)7CH2=CH2分子的对称性,两者与·OH反应分别考虑3条(1条氢夺取+2条·OH加成)和6条(3条氢夺取+3条·OH加成)反应途径,所有模型化合物与·OH反应的反应途径见图2.
表1给出了在不同方法下计算的模型化合物的kOH值. 可以看出,CF3CF2H、 CF3CH2CF3、CF3CF2(CHF)2CF3、CF2CH2和CF3CH2CF3的kOH范围分别为6.79 × 10−16 — 4.93 × 10−14 cm3·molecule−1·s−1、8.47 × 10−17 — 5.99 × 10−15 cm3·molecule−1·s−1、2.10 × 10−15 — 5.65 × 10−14 cm3·molecule−1·s−1、4.81 × 10−13 — 8.90 × 10−11 cm3·molecule−1·s−1;7.27 × 10−14 — 1.08 × 10−11 cm3·molecule−1·s−1. 它们对应的大气半减期范围分别为:0.40 — 29.00 a;3.29 — 232.83 a;0.34 — 9.39 a;0.08 — 14.76 d;0.65 — 97.66 d. 表明不同计算方法对HFCs和HFOs的kOH值和持久性评估的影响较大. 此外,HFOs的kOH值普遍大于HFCs的kOH值的研究结果表明在对流层条件下,HFOs与·OH反应更快,更容易被·OH氧化去除.
-
表2列出了HFCs和HFOs实测与计算的lgkOH平均绝对误差(MAE),当MAE值小于0.500时认为方法预测的kOH的效果较好. 对于HFCs,ωB97X-D结合κS修正的TST的效果不理想(MAE的范围为0.692 — 1.003);M06-2X-D3结合κS修正的TST方法更具优势,其MAE值均小于0.250. 其中,采用基组def2-TZVP(MAE = 0.169)、may-cc-pVTZ (MAE = 0.170)、jun-cc-pVTZ (MAE = 0.178)、def2-TZVPP (MAE = 0.182)、jul-cc-pVTZ (MAE = 0.193)和aug-cc-pVTZ (MAE = 0.197)计算方法效果更优. 因此,当计算·OH和HFCs的反应时,建议使用上述修正TST和计算单点能的方法计算HFCs的kOH值. 对于HFOs,M06-2X-D3结合aug-cc-pVTZ基组计算单点能,并采用κW修正的TST计算kOH,得到的lgkOH的MAE最小(0.497). 因此,建议使用κW修正的TST方法结合M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ计算HFOs的kOH值.
对比所有模型化合物(3个HFCs和2个HFOs)lgkOH的MAE值,发现M06-2X-D3和ωB97X-D泛函结合不同基组得到的lgkOH的MAE范围分别为0.34—0.78和0.51—1.11. κS修正的TST方法结合M06-2X-D3泛函计算单点能的MAE值范围为0.34—0.50,均≤0.500. 按照MAE值排序,利用κS修正TST的动力学方法结合不同单点能计算方法中,最优的2种方法分别为结合M06-2X-D3/aug-cc-pVTZ (MAE = 0.34)和结合M06-2X-D3/jul-cc-pVTZ (MAE = 0.35). κW修正TST的动力学方法结合不同单点能计算方法中,最优的2种方法分别为结合M06-2X-D3/aug-cc-pVTZ (MAE = 0.47)和结合M06-2X-D3/jul-cc-pVTZ (MAE = 0.49). 因此,本研究推荐κS修正的M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ或者M06-2X-D3/jul-cc-pVTZ//M06-2X/cc-pVDZ方法计算HFCs和HFOs的kOH.
-
表3为筛选出分别适用于HCFs和HFOs的计算方法、计算得到的反应的热力学和动力学参数,其中HCFs的∆V均为0 kJ·mol−1. 对于3种HFCs,可以看出·OH 夺取HFCs上的H原子时,焓变(ΔH)均小于0 kJ·mol−1,表明反应可自发进行. 然而由于Δ E较高(12.75—23.16 kJ·mol−1),在298 K条件下反应很难发生. 表明HFCs可能在大气中持久存在. 此外,对比3种HFCs (CF3CH2F、CF3CH2CF3和CF3CF2(CHF)2CF3)的kOH值,可以看出碳链长度对HFCs的kOH几乎没有影响.
对于2种HFOs,可以看出所有反应的ΔH值小于0 kJ·mol−1,表明反应是放热反应. 而H夺取反应途径的ΔE值明显高于加成反应,表明·OH加成反应是·OH与2种HFOs反应的主要反应通道. 对比动力学数据,CF2=CH2和CF3(CF2)7CH2=CH2双键加成的产物分支比分别为99.99%和99.96%,同样证明双键加成是主要的反应机制. 此外,双键加成(3d, 4d)和(5d, 6d)反应通道的kOH值分别为3.76 × 10−13 cm3 ·molecule−1·s−1和4.00 × 10−13 cm3·molecule−1·s−1,说明—CF2和—CH2对kOH的影响较小. 值得注意的是,·OH加成到CF3(CF2)7CH2=CH2双键不同位置上时,其kOH的值也有明显不同. 5e的kOH为(1.74 × 10−13 cm3·molecule−1·s−1)明显高于4e (7.76 × 10−15 cm3·molecule−1·s−1)和6e (2.04 × 10−15 cm3·molecule−1·s−1)反应通道. 5e反应通道的产物分支比为94.62%,这表明·OH更容易与CF3(CF2)7CH2=CH2以5e的反应通道反应. 如图3中4e、5e和6e的过渡态所示,·OH在加成过程中,可能受到碳链上F原子空间位阻的影响.
-
本研究以5个HFCs、HFOs的kOH实测值作为参照,从16种单点能计算方法和2种动力学计算方法中筛选适合HFCs和HFOs的kOH值的热力学和动力学参数的计算方法. 以MAE作为检验计算方法效果的标准,HFCs推荐使用κS修正TST结合M06-2X-D3/def2-TZVP//M06-2X/cc-pVDZ方法计算kOH;HFOs推荐使用κW修正TST结合M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ方法计算kOH;推荐使用κS修正TST方法结合的M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ或M06-2X-D3/jul-cc-pVTZ//M06-2X/cc-pVDZ方法计算HFCs、HFOs的kOH. 此研究筛选了适用于计算HFCs和HFOs的kOH值的量子化学方法,为高效、准确预测HFCs和HFOs的kOH和评估其大气持久性提供了方法支撑.
气相氢氟烃和氢氟烯烃与·OH反应的量子化学计算方法筛选
Screening of quantum chemical method for the reactions of hydrofluorocarbons and hydrofluoroolefins with ·OH in the Atmosphere
-
摘要: 氢氟烃 (HFCs)和氢氟烯烃 (HFOs)常被用作氢氯氟烃的替代物. 为评估HFCs和HFOs是否可以理想替代氢氯氟烃,需要对其大气转化进行充分研究,尤其需要充分了解其大气持久性的信息. 目前用于评估化学品大气持久性的重要参数气相羟基自由基(·OH)二级反应速率常数(kOH)的数据量尚不能满足多种HFCs和HFOs的评估. 因此有必要发展能够快速预测kOH的方法. 量子化学计算方法具有高效、准确的优点,是预测kOH的重要手段. 然而目前研究使用的量子化学方法纷繁复杂,亟需筛选适合HFCs和HFOs的量子化学方法. 本研究基于3种HFCs(CF3CF2H、CF3CH2CF3和CF3CF2(CHF)2CF3)和2个HFOs(CF2CH2和CF3CH2CF3)的实验数据,从多种热力学参数计算方法和动力学计算方法中筛选适用于计算HFCs和HFOs气相kOH的方法. 研究结果表明,通过对比lgkOH的实测值与不同计算方法所得计算值之间的平均绝对误差(MAE),利用Skodje-Truhlar隧道效应校正系数 (κS)修正传统过渡态理论(TST),再结合M06-2X-D3/def2-TZVP//M06-2X/cc-pVDZ水平的密度泛函理论(DFT)计算HFCs的kOH效果最好,其MAE值为0.17;采用Wigner隧道效应校正系数 (κW)修正的TST结合M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ (MAE = 0.50)的方法计算HFOs的kOH效果最好;而κS修正TST的M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ (MAE = 0.34)或M06-2X-D3/jul-cc-pVTZ//M06-2X/cc-pVDZ (MAE = 0.35)方法都适用于计算HFCs和HFOs的kOH. 本研究筛选的方法为快速、准确计算HFCs和HFOs的kOH及评估其大气持久性提供了方法支撑.
-
关键词:
- 氢氟烃 (HFCs) /
- 氢氟烯烃 (HFOs) /
- ·OH /
- 量子化学计算 /
- 密度泛函理论(DFT) /
- 动力学.
Abstract: Hydrofluorocarbons (HFCs) and hydrofluoroolefins (HFOs) are mainly employed to substitute hydrochlorofluorocarbons. In order to evaluate whether the HFCs and HFOs are ideal alternatives for hydrochlorofluorocarbons or not, it is necessary to fully explore their atmospheric transformation, especially the information atmospheric persistence. To date, the quantity of second-order reaction rate constants (kOH) for chemicals reacting with hydroxyl radicals (·OH), which are essential parameters to characterize the atmospheric persistence of HFCs and HFOs, cannot meet the needs of atmospheric persistence assessment for HFCs and HFOs. Therefore, it is necessary to develop a method that can predict the kOH values efficiently. Considering the efficiency and accuracy of quantum chemical calculation, quantum chemical calculation is an important way to predict the kOH values. However, the quantum chemistry methods used in the current research are complex, and it is urgent to screen the quantum chemistry methods that are suitable for HFCs and HFOs. In this study, suitable methods for predicting the atmospheric kOH values of HFCs and HFOs were selected from a variety of thermodynamic parameter calculation methods and kinetics calculation methods based on the experimental data of 3 HFCs (CF3CF2H, CF3CH2CF3, and CF3CF2(CHF)2CF3) and 2 HFOs (CF2CH2 and CF3CH2CF3). The research results show that by comparing the mean absolute error (MAE) between the experimental lgkOH values and the lgkOH values calculated by different theoretical methods, the method employing the traditional transition state theory (TST) modified with the Skodje-Truhlar tunnel effect correction coefficient(κS) and combining with the density functional theory (DFT) at the M06-2X-D3/def2-TZVP//M06-2X/cc-pVDZ level has the best effect on calculating the kOH of HFCs accurately, whose MAE was 0.17; The method employing TST method modified with Wigner transmission coefficient (κW) and combining with the M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ (MAE= 0.50) showed the best performance for calculating the kOH values of HFOs; Both of the two methods that TST modified with the κW correction combine with M06-2X-D3/aug-cc-pVTZ//M06-2X/cc-pVDZ (MAE = 0.34) or M06-2X-D3/jul-cc-pVTZ//M06-2X/cc-pVDZ (MAE = 0.35) were suitable for the kOH prediction of HFCs and HFOs. In this study, the selected methods provide efficient and accurate methods for the kOH calculation and atmospheric persistence assessment of HFCs and HFOs. -

-
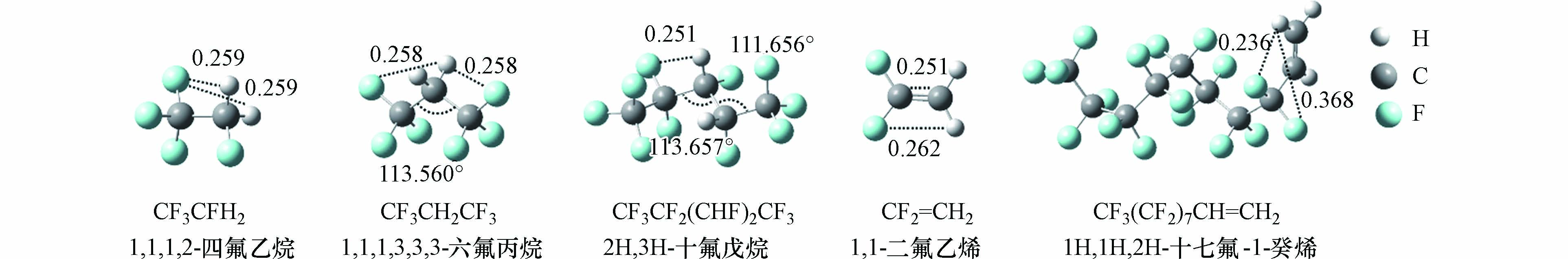
图 1 参考HFCs和HFOs的最低能量构象(距离单位为nm)
Figure 1. Global minimum conformations of HFCs and HFOs as reference (The unit of distance is nm)

图 2 CF3CFH2 (A)、CF3CH2CF3 (B)、C2F5(CHF)2CF3 (C)、 CF2=CH2 (D)、 CF3(CF2)7CH2=CH2 (E)与·OH反应的所有反应通道
Figure 2. Possible pathways for the reactions of CF3CFH2 (A), CF3CH2CF3 (B), C2F5(CHF)2CF3 (C), CF2=CH2 (D), CF3(CF2)7CH2=CH2 (E) with ·OH.
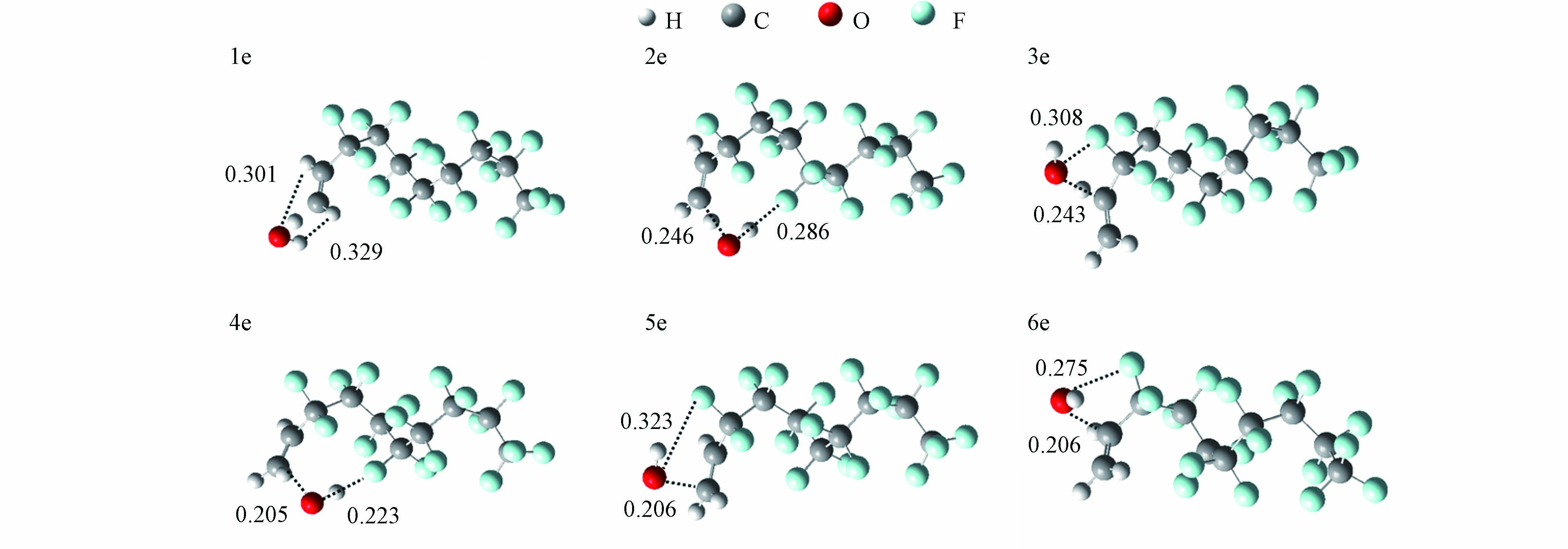
图 3 ·OH与CF3(CF2)7CH=CH2反应的过渡态结构(距离单位为nm)
Figure 3. Transition-state geometries for the reaction of ·OH with CF3(CF2)7CH=CH2 (The unit of distance is nm)
表 1 过渡态理论(TST)结合隧道效应校正 (κ)和不同单点能方法计算的模型化合物的kOH (cm3·molecule−1·s−1)
Table 1. kOH (cm3·molecule−1·s−1) of selected compounds calculated by the combination transition-state theory (TST) with different transmission coefficient (κ) correction and single-point-energy calculation methods.
单点能方法
Zero-point energy methodκ 化合物
CompoundCF3CF2H CF3CH2CF3 CF3CF2(CHF)2CF3 CF2=CH2 CF3(CF2)7CH=CH2 实测值 6.25 × 10−15 9.57 × 10−16 3.29 × 10−15 2.49 × 10−12 1.36 × 10−12 M06-2X-D3/aug-cc-pVTZ κS 4.11 × 10−15 1.16 × 10−15 8.55 × 10−15 1.58 × 10−12 1.34 × 10−13 κW 1.27 × 10−15 1.79 × 10−16 3.98 × 10−15 1.55 × 10−12 1.84 × 10−13 M06-2X-D3/may-cc-pVTZ κS 3.19 × 10−15 9.33 × 10−16 6.86 × 10−15 8.18 × 10−13 1.22 × 10−13 κW 9.22 × 10−16 1.35 × 10−16 2.96 × 10−15 1.02 × 10−12 1.48 × 10−13 M06-2X-D3/jun-cc-pVTZ κS 3.50 × 10−15 1.01 × 10−15 7.28 × 10−15 1.23× 10−12 1.24 × 10−13 κW 1.03 × 10−15 1.49 × 10−16 3.20 × 10−15 1.20 × 10−12 1.55 × 10−13 M06-2X-D3/jul-cc-pVTZ κS 3.78 × 10−15 1.10 × 10−15 8.05 × 10−15 1.51 × 10−12 1.31 × 10−13 κW 1.14 × 10−15 1.67 × 10−16 3.67 × 10−15 1.48 × 10−12 1.73 × 10−13 M06-2X-D3/def2-TZVP κS 2.96 × 10−15 9.03 × 10−16 6.51 × 10−15 8.07× 10−13 1.33 × 10−13 κW 8.40 × 10−16 1.29 × 10−16 2.75 × 10−15 9.62 × 10−13 1.88 × 10−13 M06-2X-D3/def2-TZVPP κS 4.27 × 10−15 1.12 × 10−15 8.28 × 10−15 7.25 × 10−13 1.14 × 10−13 κW 1.33 × 10−15 1.71 × 10−16 3.80 × 10−15 8.58 × 10−13 1.34 × 10−13 M06-2X-D3/pcseg-2 κS 2.50 × 10−15 6.47 × 10−16 5.31 × 10−15 4.81 × 10−13 7.27 × 10−14 κW 6.79 × 10−16 8.47 × 10−17 2.10 × 10−15 5.57 × 10−13 7.52 × 10−14 M06-2X-D3/MG3S κS 4.16 × 10−15 1.18 × 10−15 1.18 × 10−14 1.59 × 10−12 1.32 × 10−13 κW 1.29 × 10−15 1.82 × 10−16 6.08 × 10−15 1.56 × 10−12 1.71 × 10−13 ωB97X-D/aug-cc-pVTZ κS 3.99 × 10−14 4.43 × 10−15 3.45 × 10−14 5.10 × 10−11 4.72 × 10−12 κW 2.72 × 10−14 1.08 × 10−15 2.96 × 10−14 4.99 × 10−11 4.54 × 10−12 ωB97X-D/may-cc-pVTZ κS 3.34 × 10−14 3.65 × 10−15 2.92 × 10−14 3.51 × 10−11 3.84 × 10−12 κW 2.08 × 10−14 8.32 × 10−16 2.26 × 10−14 3.44 × 10−11 3.69 × 10−12 ωB97X-D/jun-cc-pVTZ κS 3.57 × 10−14 3.97 × 10−15 3.09 × 10−14 4.17 × 10−11 4.10 × 10−12 κW 2.30 × 10−14 9.34 × 10−16 2.48 × 10−14 4.11 × 10−11 3.94 × 10−12 ωB97X-D/jul-cc-pVTZ κS 3.75 × 10−14 4.24 × 10−15 3.31 × 10−14 4.88 × 10−11 4.51 × 10−12 κW 2.47 × 10−14 1.02 × 10−15 2.77× 10−14 4.78 × 10−11 4.34 × 10−12 ωB97X-D/def2-TZVP κS 3.43 × 10−14 4.49 × 10−15 3.27 × 10−14 2.94 × 10−11 5.78 × 10−12 κW 2.17 × 10−14 1.09 × 10−15 2.71 × 10−14 2.88 × 10−11 5.56 × 10−12 ωB97X-D/def2-TZVPP κS 4.29 × 10−14 5.29 × 10−15 3.70 × 10−14 2.99 × 10−11 4.56 × 10−12 κW 3.07 × 10−14 1.36 × 10−15 3.33 × 10−14 2.93 × 10−11 4.38 × 10−12 ωB97X-D/pcseg-2 κS 2.53 × 10−14 2.93 × 10−15 2.35 × 10−14 2.06 × 10−11 2.58 × 10−12 κW 1.40 × 10−14 6.24 × 10−16 1.63 × 10−14 2.01 × 10−11 2.53 × 10−12 ωB97X-D/MG3S κS 4.93× 10−14 5.99 × 10−15 5.07 × 10−14 8.90 × 10−11 1.08 × 10−11 κW 3.75 × 10−14 1.59 × 10−15 5.65 × 10−14 8.71 × 10−11 1.04 × 10−11  下载: 导出CSV
下载: 导出CSV
表 2 理论计算lgkOH的平均绝对误差(MAE) (kOH: cm3·molecule−1·s−1)
Table 2. Mean absolute deviation (MAE) values of theoretical lgkOH for selected compounds. (kOH: cm3·molecule−1·s−1)
单点能方法
Zero-point energy methodMAE (TST × κW) MAE (TST × κS) HFCs和HFCs HFCs HFOs HFCs和HFCs HFCs HFOs M06-2X-D3/aug-cc-pVTZ 0.47 0.45 0.50 0.34 0.20 0.56 M06-2X-D3/may-cc-pVTZ 0.57 0.53 0.64 0.39 0.17 0.72 M06-2X-D3/jun-cc-pVTZ 0.53 0.49 0.59 0.36 0.18 0.63 M06-2X-D3/jul-cc-pVTZ 0.49 0.46 0.52 0.35 0.19 0.58 M06-2X-D3/def2-TZVP 0.58 0.56 0.60 0.38 0.17 0.69 M06-2X-D3/def2-TZVPP 0.54 0.44 0.69 0.41 0.18 0.75 M06-2X-D3/pcseg-2 0.78 0.69 0.91 0.50 0.21 0.93 M06-2X-D3/MG3S 0.51 0.51 0.51 0.37 0.24 0.56 ωB97x-D/aug-cc-pVTZ 0.74 0.59 0.95 0.91 0.87 0.97 ωB97X-D/may-cc-pVTZ 0.63 0.50 0.83 0.81 0.80 0.84 ωB97X-D/jun-cc-pVTZ 0.66 0.52 0.88 0.85 0.83 0.89 ωB97X-D/jul-cc-pVTZ 0.71 0.56 0.93 0.89 0.85 0.95 ωB97X-D/def2-TZVP 0.68 0.55 0.88 0.86 0.85 0.89 ωB97X-D/def2-TZVPP 0.73 0.66 0.83 0.89 0.92 0.84 ωB97X-D/pcseg-2 0.51 0.43 0.63 0.67 0.69 0.64 ωB97X-D/MG3S 0.97 0.79 1.25 1.11 1.00 1.27
下载: 导出CSV
表 3 模型化合物与·OH反应的能垒(ΔE: kJ·mol−1, 0 K)、标准活化自由能(ΔG‡,0: kJ·mol−1, 298 K)、反应焓变(ΔrH: kJ·mol−1, 298 K)、过渡态虚频(υi†: cm−1)和kOH (cm3·molecule−1 s−1, 298 K)的计算值
Table 3. Calculated energy barrier (ΔE: kJ·mol−1, 0 K), the standard Gibbs free energy of activation (ΔG‡,0: kJ·mol−1, 298 K), enthalpy (ΔH: kJ·mol−1, 298 K), frequency of TSs (υi†: cm−1) and kOH (cm3·molecule−1·s−1, 298 K) values for selected compounds reacting with ·OH.
反应通道
Reaction pathwayκ 单点能方法
Zero-point energy methodΔG‡,0 ∆E ΔH υi† kOH CF3CH2F 实测kOH:6.25 × 10−15 计算kOH:2.96 × 10−15 1a, 2a κS M06-2X-D3/def2-TZVP 52.90 17.71 −62.67 1461.49 1.48 × 10−15 CF3CH2CF3 实测kOH:9.57 × 10−16 计算kOH:9.03× 10−16 1b κS M06-2X-D3/def2-TZVP 58.07 23.16 −46.48 1575.73 4.00 × 10−16 2b κS M06-2X-D3/def2-TZVP 57.61 22.83 −43.77 1600.20 5.03 × 10−16 CF3CF2(CHF)2CF3 实测kOH:3.29 × 10−15 计算kOH:6.51 × 10−15 1c κS M06-2X-D3/def2-TZVP 52.08 15.42 −73.12 1486.68 1.79 × 10−15 2c M06-2X-D3/def2-TZVP 48.77 12.75 −74.71 1429.22 4.73 × 10−15 CF2=CH2 实测kOH:2.49 × 10−12 计算kOH:1.55 × 10−12 1d, 2d κW M06-2X-D3/aug-cc-pVTZ 67.08 8.22 −3.11 1344.44 1.23 × 10−18 3d, 4d κW M06-2X-D3/aug-cc-pVTZ 33.54 −0.15 −128.79 437.72 4.00 × 10−13 5d, 6d κW M06-2X-D3/aug-cc-pVTZ 33.62 −0.64 −186.12 392.38 3.76 × 10−13 CF3(CF2)7CH=CH2 实测kOH:1.36 × 10−12 计算kOH:1.84 × 10−13 1e κW M06-2X-D3/aug-cc-pVTZ 59.06 25.11 −25.63 1532.35 3.73 × 10−17 2e κW M06-2X-D3/aug-cc-pVTZ 61.97 25.33 −24.34 1619.52 1.25 × 10−17 3e κW M06-2X-D3/aug-cc-pVTZ 60.40 25.27 −23.02 1527.16 2.16 × 10−17 4e κW M06-2X-D3/aug-cc-pVTZ 43.50 6.34 −129.20 536.44 7.76 × 10−15 5e κW M06-2X-D3/aug-cc-pVTZ 35.72 0.64 −127.38 497.65 1.74 × 10−13 6e κW M06-2X-D3/aug-cc-pVTZ 46.68 8.99 −118.41 470.14 2.04 × 10−15
下载: 导出CSV
-
[1] HODNEBROG, ETMINAN M, FUGLESTVEDT J S, et al. Global warming potentials and radiative efficiencies of halocarbons and related compounds: A comprehensive review [J]. Reviews of Geophysics, 2013, 51(2): 300-378. doi: 10.1002/rog.20013 [2] ABAS N, KALAIR A R, KHAN N, et al. Natural and synthetic refrigerants, global warming: A review [J]. Renewable and Sustainable Energy Reviews, 2018, 90: 557-569. doi: 10.1016/j.rser.2018.03.099 [3] BIRMPILI T. Montreal protocol at 30: The governance structure, the evolution, and the Kigali amendment [J]. Comptes Rendus Geoscience, 2018, 350(7): 425-431. doi: 10.1016/j.crte.2018.09.002 [4] FLERLAGE H, VELDERS G J M, de BOER J. A review of bottom-up and top-down emission estimates of hydrofluorocarbons (HFCs) in different parts of the world [J]. Chemosphere, 2021, 283: 131208. doi: 10.1016/j.chemosphere.2021.131208 [5] YI L Y, WU J, AN M D, et al. The atmospheric concentrations and emissions of major halocarbons in China during 2009-2019 [J]. Environmental Pollution, 2021, 284: 117190. doi: 10.1016/j.envpol.2021.117190 [6] GLIGOROVSKI S, STREKOWSKI R, BARBATI S, et al. Environmental implications of hydroxyl radicals ((•)OH) [J]. Chemical Reviews, 2015, 115(24): 13051-13092. doi: 10.1021/cr500310b [7] SUN X Y, HU Y M, XU F, et al. Mechanism and kinetic studies for OH radical-initiated atmospheric oxidation of methyl propionate [J]. Atmospheric Environment, 2012, 63: 14-21. doi: 10.1016/j.atmosenv.2012.08.045 [8] LIAO Z H, ZENG M, WANG L M. Atmospheric oxidation mechansim of polychlorinated biphenyls (PCBs) initiated by OH radicals [J]. Chemosphere, 2020, 240: 124756. doi: 10.1016/j.chemosphere.2019.124756 [9] SHI X L, ZHANG R M, LI Y F, et al. Mechanism theoretical study on OH-initiated atmospheric oxidation degradation of dimethoate [J]. Journal of Molecular Structure, 2018, 1163: 61-67. doi: 10.1016/j.molstruc.2018.02.104 [10] HOLTOMO O, NGUE'ZEO H, NSANGOU M, et al. Theoretical investigation of the atmospheric implication for the reaction of •OH radical with CF2C(CH3)-CX3, X = H, F [J]. Journal of Molecular Graphics and Modelling, 2021, 106: 107905. doi: 10.1016/j.jmgm.2021.107905 [11] GUPTA P, RAJAKUMAR B. A theoretical insight on the kinetics for the reaction of (E)-/ (Z)-CHF=CF(CF2)x=1, 2CF3 with OH radicals under tropospheric conditions [J]. Journal of Fluorine Chemistry, 2019, 222/223: 31-45. doi: 10.1016/j.jfluchem.2019.04.009 [12] JABEEN F, KUMAR A, RAJAKUMAR B. Kinetics, thermochemistry and atmospheric implications for the reaction of OH radicals with CH3CF = CF2 (HFO-1243yc) [J]. Chemical Physics Letters, 2020, 758: 137933. doi: 10.1016/j.cplett.2020.137933 [13] GOGOI P, PAUL S, MISHRA B K, et al. Tropospheric oxidation of 1H-heptafluorocyclopentene (cyc-CF2CF2CF2CF═CH–) with OH radicals: Reaction mechanism, kinetics, and global warming potentials [J]. ACS Earth and Space Chemistry, 2021, 5(7): 1792-1800. doi: 10.1021/acsearthspacechem.1c00124 [14] XU C, WANG C Y, LI B, et al. Theoretical study on the reaction mechanism of OH radical with Z(E)-CF3CH CHF [J]. Physical Chemistry Chemical Physics, 2019, 21(3): 1367-1374. doi: 10.1039/C8CP06647G [15] HSU K J, DEMORE W B. Rate constants and temperature dependences for the reactions of hydroxyl radical with several halogenated methanes, ethanes, and propanes by relative rate measurements [J]. The Journal of Physical Chemistry, 1995, 99(4): 1235-1244. doi: 10.1021/j100004a025 [16] CHEN L, UCHIMARU T, KUTSUNA S, et al. Kinetics study of gas-phase reactions of erythro/threo-CF3CHFCHFC2F5 with OH radicals at 253-328 K [J]. Chemical Physics Letters, 2010, 488(1/2/3): 22-26. [17] TOKUHASHI K, TAKIZAWA K, KONDO S. Rate constants for the reactions of OH radicals with fluorinated ethenes: Kinetic measurements and correlation between structure and reactivity [J]. The Journal of Physical Chemistry. A, 2018, 122(19): 4593-4600. doi: 10.1021/acs.jpca.7b11653 [18] ANDERSEN M P S, NIELSEN O J, TOFT A, et al. Atmospheric chemistry of CxF2x + 1CHCH2 (x = 1, 2, 4, 6, and 8): Kinetics of gas-phase reactions with Cl atoms, OH radicals, and O3 [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2005, 176(1/2/3): 124-128. [19] SUTCLIFFE B T, WOOLLEY R G. On the quantum theory of molecules [J]. The Journal of Chemical Physics, 2012, 137(22): 22A-544A. [20] KÜHNE T D, IANNUZZI M, del BEN M, et al. CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations [J]. The Journal of Chemical Physics, 2020, 152(19): 194103. doi: 10.1063/5.0007045 [21] ZHAO Y, TRUHLAR D G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals [J]. Theoretical Chemistry Accounts, 2008, 120(1): 215-241. [22] DUNNING T H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen [J]. The Journal of Chemical Physics, 1989, 90(2): 1007-1023. doi: 10.1063/1.456153 [23] FRISCH M, TRUCKS G, SCHLEGEL H, et al. Gaussian 09, rev[CP]. Gaussian Inc, Wallingford, 2009 [24] LI C, XIE H B, CHEN J W, et al. Predicting gaseous reaction rates of short chain chlorinated paraffins with ·OH: Overcoming the difficulty in experimental determination [J]. Environmental Science & Technology, 2014, 48(23): 13808-13816. [25] CHAI J D, HEAD-GORDON M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections [J]. Physical Chemistry Chemical Physics, 2008, 10(44): 6615-6620. doi: 10.1039/b810189b [26] WEIGEND F. Accurate Coulomb-fitting basis sets for H to Rn [J]. Physical Chemistry Chemical Physics, 2006, 8(9): 1057-1065. doi: 10.1039/b515623h [27] FERNANDEZ-RAMOS A, ELLINGSON B A, GARRETT B C, et al. Variational transition state theory with multidimensional tunneling[M]//Reviews in Computational Chemistry. Hoboken, NJ, USA: John Wiley & Sons, Inc. , 2007: 125-232. [28] WIGNER E. The transition state method [J]. Transactions of the Faraday Society, 1938, 34: 29-41. doi: 10.1039/tf9383400029 [29] DOUBLEDAY C, ARMAS R, WALKER D, et al. Heavy-atom tunneling calculations in thirteen organic reactions: Tunneling contributions are substantial, and Bell's formula closely approximates multidimensional tunneling at ≥250 K [J]. Angewandte Chemie (International Ed. in English), 2017, 56(42): 13099-13102. doi: 10.1002/anie.201708489 [30] SKODJE R T, TRUHLAR D G. Parabolic tunneling calculations [J]. The Journal of Physical Chemistry, 1981, 85(6): 624-628. doi: 10.1021/j150606a003 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 2032
- HTML全文浏览数: 2032
- PDF下载数: 66
- 施引文献: 0
