

-
对硝基苯酚等芳香族酚类化合物是合成有机化学工业废水中常见的高毒性、强致癌、耐生物降解有机污染物[1].此外,不少芳香族化合物作为有机染料广泛应用于消费品制造业中,具有较高毒性、水溶性和稳定性,自然降解速度相当缓慢,未经处理排入水体会对环境和人体造成持续危害[2].目前已开发出包括吸附法、萃取法、膜分离法、微生物降解法、高级氧化法等多种处理该类污染水体的方法,但存在二次污染、操作繁琐、设备复杂、处理成本高等问题[3-4].特别地,将硝基苯类物质还原为胺类物质是非常重要的化学资源转化,后者是许多有较高经济价值的化学品(如解热镇痛药、农药、炸药、染料、润滑剂等)的必要成分,而且氨基官能团常用作衍生合成最终化学品的关键位点[1,5].通过活性金属固载的纳米催化剂非均相催化还原工艺将水体中高毒性有机污染物转化为低毒性、高附加值且可重复使用的化学物质,不仅能促进工业有机废水资源转化和综合利用,降低环境风险,而且方法简单、成本较低、易于获取,是消除该类污染水体极其高效、绿色且经济的技术方法[5-7].
近年来,应用于还原降解芳香族类污染物的高效载金纳米催化剂的研发热度持续不减,也取得了显著进展,但保持纳米Au催化剂的高活性和高稳定性仍然是难度较大的瓶颈[7-10].纳米Au颗粒的制备粒径越小,其原子催化效率和使用效益愈发提升,但由此呈现的Au颗粒表面能越高,热力学稳定性越差,容易发生团聚和变形,严重限制其大规模催化应用[8-10].因此,通过设计特定的载金纳米结构来获得高活性、稳定性纳米催化剂是提高其整体催化性能和使用寿命的可行策略.利用多孔载体对纳米Au颗粒进行物理空间分割实现固载和分散,可一定程度限制Au颗粒的聚集[7-11].或者,利用金属氧化物或第二种金属晶体特殊化学界面效应对纳米Au颗粒进行化学修饰形成异质结构或双金属合金得到有效稳定[12-15].结合上述策略,核壳复合材料可作为理想载体进行特定功能化载金催化剂新型纳米结构的设计:①可调控的核/壳双层结构能作为支撑载体锚定纳米Au颗粒的空间分布位置[14-15];②核/壳双层微观结构的多功能多组分设计可赋予该核壳结构新材料特定的物理化学性质和功能属性,以满足高效催化体系的特定应用需求[14-18].
介孔碳空心球结构广泛应用于吸附、催化和能量存储,在制备核壳复合材料中具有特殊优势,包括低密度、电子导电性、稳定性和疏水性等[18-20].与SiO2制备过程相似,间苯二酚-甲醛树脂(RF)可通过扩展经典Stöber法的应用实现聚合物层的合成与沉积.该Stöber反应过程中,乙醇-水作为溶液体系,氨水作为碱性催化剂,间苯二酚-甲醛聚合反应和硅酸四乙酯缩合反应存在明显的速率差异,因而在同一个反应体系中可实现SiO2@RF叠层复合物的合成[19-20].通过碳化热解,碳前驱体RF树脂形成介孔碳衍生壳层,SiO2硬模板被碱液刻蚀形成内空腔.该合成路线可实现介孔碳为壳层的载金核壳结构.此外,对内核结构进行磁性组分的整合以构筑具有较好磁回收性能的载金核壳复合材料,可提高纳米Au催化剂分离回收效率和循环使用性能,有效缓解金材料供应有限且成本昂贵的不利因素[16,18-19].
本文采用乙二胺介导的Stöber-SiO2/RF扩展工艺及碳化-水热刻蚀法制备具有介孔碳氮壳层的Fe2O3@CeO2/CN核壳椭球;[Au(en)2]3+为金前驱体固载于上述核壳结构,通过还原气氛热处理转化为较好分散度的纳米Au颗粒,Fe2O3内核同步转化为小体积Fe颗粒,形成内空腔,得到Fe@CeO2-Au/CN双空腔核壳磁性椭球催化剂.对其进行微观结构和复合组成表征分析,研究其催化还原对硝基苯酚和典型染料分子的反应性能和循环使用性能,探讨催化剂独特结构特征和复合组成协同效应对其催化反应性能的作用影响,揭示其催化反应机理,为设计该类污染水体深度处理的高活性、稳定性催化剂以及提升催化还原工艺的实践效果提供可借鉴的方法.
-
首先配置80 mL含有0.025 mol·L−1 FeCl3和0.48 mmol·L−1 NaH2PO4的水溶液,转入反应釜,105℃水热反应48 h,得到Fe2O3梭型微粒;称取烘干的0.2 g Fe2O3固体粉末和0.75 g聚乙烯吡咯烷酮(K-30)超声分散于40 mL乙醇,同时称取0.5 g Ce(NO3)3·6H2O和0.9 g六亚甲基四胺超声溶解于60 mL去离子水,混合上述两种混合物并搅拌30 min,随后升温至75℃进行回流反应3 h;降至室温后,离心回收反应产物,用乙醇洗涤,烘干,得到Fe2O3@CeO2微粒.
-
取0.15 g Fe2O3@CeO2固体、0.08 mL乙二胺、0.4 g十六烷基三甲基氯化铵、20 mL乙醇超声分散于50 mL去离子水中,随后升温至70℃搅拌混合,加入0.2 g间苯二酚,30 min后加入0.8 g硅酸四乙酯和0.2 mL甲醛,搅拌反应24 h;降至室温后,离心回收反应产物,用乙醇洗涤,烘干,得到Fe2O3@CeO2@SR核壳椭球.
-
取上述Fe2O3@CeO2@SR椭球在高纯氮气氛围进行碳化处理,碳化条件为:以5℃·min−1升温速率加热到150℃,保温1 h,再加热到500℃保持2 h;然后将碳化后的椭球超声分散到5%氨水溶液中,转入反应釜,150℃水热反应6 h;降至室温后,离心回收反应产物,用去离子水洗涤,烘干,得到Fe2O3@CeO2/CN核壳椭球.
-
配制0.2 mol·L−1的HAuCl4溶液,按6% wt比例滴加适量的乙二胺,搅拌反应1 h,得到棕色溶胶,然后向其中按比例加进乙醇连续混合2 h,离心回收白色沉淀物,用去离子水清洗,40℃真空干燥12 h,得到淡黄色Au(en)2Cl3粉末;称取0.1 g Au(en)2Cl3粉末溶解于60 mL去离子水中,混合状态下滴加NaOH溶液,调节溶液pH为10,得到淡黄色Au(en)2Cl3溶液,放置冰箱备用;称取20 mg Fe2O3@CeO2/CN核壳椭球超声分散于10 mL Au(en)2Cl3溶液,混合反应24 h,离心回收沉淀物,多次洗涤,40℃真空烘干,得到乙二胺金改性中间物Fe2O3@CeO2-AE/CN;调制高纯氢气焙烧氛围,以5℃·min−1为升温速率,升温至500℃保持2 h,得到Fe@CeO2-Au/CN双空腔核壳磁性椭球.
-
TEM、HRTEM和EDS谱图采用FEI Tecnai G20透射电镜观察获得. SEM图采用Zeiss (Ultra Plus)扫描电镜观察获得.晶相结构XRD谱图通过XRD SmartLab(Rigaku)衍射仪采集测定,扫描角度为10°—80°.红外FTIR图采用KBr压片法通过Bruker Alpha红外光谱仪进行采集.N2吸脱附等温线在Micromeritics ASAP 2020吸脱附测定仪进行采集,样品比表面积通过BET法计算得到,孔径分布曲线采用脱附分支曲线通过BJH法计算得到.磁滞回线采用Lake Shore-7304振动样品磁强计(VSM)测定.UV-Vis DRS图通过采用UV-Vis分光光度仪(UV-3600,Shimadzu)进行测定采集.XPS谱图通过采用ESCALab220i-XL电子能谱仪进行测定采集.Raman谱图采用Renishaw inVia拉曼显微镜进行观察测定.
-
对硝基苯酚(4-NP)选择性还原降解实验具体操作如下:在锥形瓶中分别加进100 mL去离子水、5 mL对硝基苯酚溶液(0.02 mol·L−1)和20 mL NaBH4溶液(0.2 mol·L−1)保持振荡混合,向上述混合溶液中加进5 mL催化剂分散液(1 g·L−1),并对该催化反应进行计时;在不同反应时间取少量反应溶液经滤头过滤后转入比色皿,利用UV-Vis分光光度仪分析对硝基苯酚转化率.对于有机染料降解,选择初始浓度分别为0.26 mmol·L−1和0.51 mmol·L−1的亚甲基蓝(MB)和甲基橙(MO)作为底物.
-
图1为Fe@CeO2-Au/CN双空腔核壳磁性椭球催化剂的制备路线,首先水热合成Fe2O3梭型粒子内核,通过溶胶凝胶反应表面构筑CeO2颗粒层;随后采用乙二胺介导的Stöber-SiO2/RF扩展反应,利用间苯二酚-甲醛聚合反应和硅酸四乙酯缩合反应的速率差异,在Fe2O3@CeO2表面合成SiO2@RF叠层复合物(即Fe2O3@CeO2@SR),经过碳化-水热蚀刻得到具有介孔碳氮壳层和夹层空腔的Fe2O3@CeO2/CN核壳椭球.以[Au(en)2]3+为金前驱体,采用沉积沉淀反应及还原气氛热处理法在上述椭球核壳界面构筑较好分散度超细纳米Au颗粒,同时Fe2O3转化为小体积Fe颗粒并形成内空腔,Au粒径尺寸区间维持在超细尺寸范围并较好地封装固化于Fe@CeO2/CN双空腔核壳磁性复合结构中,实现纳米Au活性位的原位制备和内核磁性能的整合,得到具有独特双空腔和优良磁回收性能的Fe@CeO2-Au/CN核壳磁性椭球催化剂.其中乙二胺作为碱性催化剂和氮源,以及Au颗粒的稳定剂参与完成催化剂整体结构的制备.
-
采用TEM跟踪观察合成过程中不同样品的形貌特征和结构尺寸变化.Fe2O3梭型粒子用作形状导向模板制备双空腔核壳磁性椭球结构.图2a显示Fe2O3表面沉积CeO2颗粒层,复合物依旧呈现高长径比的单分散梭型结构,长度约大于400 nm.图2b显示Fe2O3@CeO2表面形成叠层复合壳层,原因为乙二胺介导的Stöber扩展反应下间苯二酚-甲醛聚合反应与硅酸四乙酯缩合反应存在速率差异,Fe2O3@CeO2微粒表面首先沉积SiO2层,沉积RF聚合物层,形成SiO2@RF叠层复合物,得到Fe2O3@CeO2@SR椭球.图2c显示,经过碳化-水热刻蚀处理,SiO2层被溶解形成夹层空腔,外层RF聚合物形成介孔碳氮结构,得到内核可移动的Fe2O3@CeO2/CN椭球.随后,乙二胺和HAuCl4反应合成Au(en)2Cl3前驱物,通过沉积沉淀反应对Fe2O3@CeO2/CN椭球进行乙二胺修饰和Au离子固载,实现将高含量粒径约为2 nm的Au颗粒封装在已调制好的核壳纳米结构中.其中,乙二胺作为配体,可取代[AuCl4]-中氯配体,与Au3+反应得到稳定的乙二胺金配合物[Au(en)2]3+,在碱性条件下可以稳定固定在带负电表面的氧化物载体上[21].图2d观察到大量致密的纳米Au颗粒均匀分布于CeO2/CN核壳结构,Fe颗粒如豌豆状排列并形成内核空腔,与夹层空腔形成双空腔核壳结构.
图3a的HRTEM高倍图显示Fe@CeO2-Au/CN椭球中Au颗粒(111)晶面条纹和CeO2颗粒(111)晶面条纹,图3b的SEAD图显示Fe@CeO2-Au/CN椭球具有Au、CeO2和Fe相应晶面特征斑点衍射环.图4a的SEM图显示Fe@CeO2-Au/CN椭球具有显著的双空腔核壳椭球结构,Fe内核以豌豆状颗粒排布.图4b EDS谱图中出现Fe@CeO2-Au/CN微球含有Au、Fe、Ce、O、N和C元素的峰,分别对应3.9%、26.8%、37.5%、7.8%、3.6%和20.4%的质量比.
图5a为Fe@CeO2-Au/CN椭球的XRD图,显示其分布的晶相和组分特征. 出现Fe晶相(JCPDS No. 50-1275)的特征峰,说明经过还原气氛热处理后,Fe2O3晶相转化为Fe晶相,Fe2O3对应特征峰消失.图5a还有立方萤石CeO2晶相(JCPDS No. 34-0394)的特征峰.催化剂中负载的Au颗粒晶相在2θ约为38.2°处呈现面心立方Au晶相(JCPDS No. 65-2870)在(111)晶格面的衍射峰.图5b为Fe@CeO2-Au/CN椭球的FTIR图,显示其分布的化学组成.800 cm−1对应Si—O—Si的对称伸缩振动,该位置没有红外峰,说明SiO2组分已经被刻蚀.2980—2900 cm−1位置有微弱的红外峰,对应—CH2—基团的伸缩振动,说明乙二胺作为金前驱物配体和聚合物反应介导物质修饰该椭球核壳结构中.1631 cm−1和1397 cm−1位置的红外峰为碳化处理后的CN介孔壳层中C=C和芳烃基团的特征伸缩振动.554 cm−1对应Fe—O的伸缩振动,但该位置没有出现红外峰,说明Fe@CeO2-Au/CN椭球中内核主要为Fe,已经不存在Fe2O3组分.
图6a磁滞回线显示Fe@CeO2-Au/CN椭球的饱和磁化强度为63.2 emu·g−1,说明该样品具有强磁性和高饱和磁化强度,为典型的超顺磁特质.在外加磁场的作用下,分散于溶液中的Fe@CeO2-Au/CN黑色样品快速富集至容器内壁中外加磁场一侧,原先浑浊的溶液变得澄清.Fe@CeO2-Au/CN椭球优异的磁响应性能使其在液相催化反应体系中可利用外磁场进行富集和分离,从而提高催化剂的废水处理效率以及材料利用效率.此外,Fe内核表面包覆CeO2壳层,能有效降低其磁性能损失,而且以NaBH4为还原剂的催化体系能够抑制Fe内核在催化反应过程中受到的氧化腐蚀影响.
图6b显示Fe@CeO2-Au/CN椭球具有H1型滞后环的IV型N2吸脱附曲线,为介孔结构特性.采用BJH法计算解吸分支的孔径尺寸分布曲线显示其双峰孔径分布,为双空腔介孔核壳结构特征. 孔径较小的峰对应介孔壳层结构,孔径较大的峰对应壳内空腔结构.经计算,Fe@CeO2-Au/CN椭球的BET比表面积为132 m2·g−1,孔容为0.356 cm3·g−1,平均孔径为3.27 nm.
图7a的拉曼谱图显示,Fe@CeO2-Au/CN椭球相比Fe2O3@CeO2@SR椭球在1314 cm−1和1592 cm−1出现较强的吸收峰,分别对应为CN介孔壳层中无序带(D)和石墨带(G)的吸收峰,相应的ID/IG强度比为1.08(>1),表明Fe@CeO2-Au/CN椭球中CN壳层sp2结构域分布密度和界面无序程度显著增加[22].这是由于聚合物壳层在氮气氛围高温碳化过程中转化为石墨相碳材料以及乙二胺掺杂和Au颗粒固载所致.图7b的UV-Vis漫反射图中525 nm位置出现吸收峰,为Fe@CeO2-Au/CN微球中Au颗粒表面等离子体共振模式激发下的特征吸收峰.大概在740 nm位置出现一个吸收峰,为CeO2颗粒层内Fe物种中d-d带特征吸收峰.
采用XPS分析Fe@CeO2-Au/CN椭球的表面化学态.XPS全谱中,除了Fe元素,Au、Ce、C、O和N元素都能明显被测定出来.图8a的C 1s谱图显示该微球主要以芳香环的C—C/C=C化学态为主,为碳化后的CN介孔壳层的高碳物种.图8b的N 1s谱图显示C-N的化学态特征峰,主要为掺杂于碳结构以及稳定Au颗粒的乙二胺组分.图8c的Ce 3d谱图中,在900.2 eV、897.8 eV、885.4 eV和881.6 eV的结合能处有4个峰,对应于Ce 3d5/2;而在916.2 eV和903.4 eV处的2个峰属于Ce 3d3/2.其中,897.8 eV和916.2 eV为与Ce4+相关的峰,881.6 eV和900.2 eV为与Ce3+相关的峰,其他为卫星峰,表明制备的CeO2颗粒层存在氧空位.图8d的Au 4f谱图在84.1 eV和87.8 eV处出现两个独立的峰,分别对应Au晶体在4f7/2和4f5/2结合能的特征峰.
-
选用对硝基苯酚以及典型有机染料(以亚甲基蓝和甲基橙为代表)的催化还原反应作为探针,系统探究Fe@CeO2-Au/CN椭球催化剂的独特结构设计与其催化反应性能间内在构效关系.纳米Au催化剂在温和条件下能催化还原上述污染物,被认为是消除该类污染物环境危害的成本较低、操作简单且较为环保的非均相催化方法.紫外-可见吸收光谱实时监测催化还原反应过程污染物吸收峰随时间的变化过程.对硝基苯酚本身不能被NaBH4单独还原,注入Fe@CeO2-Au/CN催化剂分散液后,对硝基苯酚溶液由亮黄色迅速褪色,5 min内反应完成.图9a显示,最终对硝基苯酚在400 nm的吸收峰完全消失,伴随产生对氨基苯酚的吸收峰.对于亚甲基蓝和甲基橙污染物,Fe@CeO2-Au/CN同样表现出较高的催化活性.图9b和图9c显示亚甲基蓝和甲基橙溶液均在6 min内迅速完成脱色,其特征吸收峰明显下降并迅速消失.这些结果充分说明Fe@CeO2-Au/CN椭球催化剂具有出色的催化还原降解对硝基苯酚和有机染料的性能.
为了进一步说明Fe@CeO2-Au/CN椭球催化剂在催化活性和稳定性方面的独特结构优势,本文采用常规方法制备了Fe@SiO2-Au/CN、Fe-Au/CN、Fe@CeO2-Au/SiO2、Fe@CeO2-Au和Fe@CeO2/CN材料作为对照样品.如图9d所示,结合不同样品的催化反应过程,计算出相应的对硝基苯酚转化率,得出Fe@CeO2-Au/CN催化性能明显优于上述对照样品.Fe@CeO2/CN几乎没有催化活性,但有19.8%的对硝基苯酚吸附去除效率,说明该椭球双空腔核壳结构高的比表面积使其具有较好的吸附性能,纳米Au颗粒是主要的催化活性组分.目标催化剂Fe@CeO2-Au/CN椭球在反应6 min内能去除99.1%的对硝基苯酚.将CN壳层替换为介孔SiO2层,得到的Fe@CeO2-Au/SiO2椭球能在7 min内消除98.2%的对硝基苯酚,说明CN介孔碳结构相比惰性SiO2能一定程度上促进固载的Au活性位的催化性能.若将CeO2内层替换为SiO2层,得到的Fe@SiO2-Au/CN椭球在8 min内消除97.6%的对硝基苯酚.若没有CeO2层作为内支撑载体,得到的Fe-Au/CN椭球在7 min内消除98.3%的对硝基苯酚.若没有CN层作为壳层保护,得到Au颗粒裸露的Fe@CeO2-Au椭球在8 min内消除98.2%的对硝基苯酚.上述结果说明Fe@CeO2-Au/CN椭球中CeO2/Au内层与CN/Au壳层形成复合协同增强作用,同时还存在CeO2与Fe协同增强作用,从而提高该双空腔载金核壳磁性椭球的催化效率;CeO2内层和Fe内核作为活性金属基体组分在构建高性能催化体系中发挥了积极作用.CN壳层有利于Fe@CeO2-Au/CN催化性能的提升,其原因为:①通过碳化乙二胺掺杂间苯二酚单体产生的π-π堆叠作用获得疏水性碳氮结构界面,增强其吸附能力,显著提高了反应进度;②介孔碳氮壳层双空腔结构加快反应物和产物扩散,强化微反应环境的特殊约束作用,使瞬时浓度较高的反应物容易到达催化剂活性位点,使反应效率更快;③碳氮结构提供促进导电的驱动力,通过改变Au表面电子态与Au颗粒发生强烈相互作用,加快Au表面在BH4-到对硝基苯酚中硝基位点上的电子迁移率,获得更高催化性能.
图9e对不同样品的ln (Ct/Co)与反应时间t进行线性拟合关联,接近于1的相关系数说明催化剂催化还原对硝基苯酚的反应过程可用一级反应动力学模型进行描述.根据动力学方程ln(Ct/Co) = -kt,由直线斜率确定的反应速率常数k对应Fe@CeO2-Au/CN、Fe@SiO2-Au/CN、Fe-Au/CN、Fe@CeO2-Au/SiO2、Fe@CeO2-Au和Fe@CeO2/CN不同样品分别为0.790、0.529、0.681、0.650、0.592、0.030 min-1,表明Fe@CeO2-Au/CN椭球催化剂的催化反应效率优于其他对照样品.由于负载型纳米Au催化剂的催化活性主要取决于Au颗粒粒径、负载含量、稳定性和金属-载体间相互作用,因此计算催化剂的TOF转换频率来反映其内在催化活性,通过将还原的对硝基苯酚摩尔量除以负载的Au摩尔量和反应时间可得到该TOF值[23].如表1所示,相比于文献报道的载金催化剂,本文制备的Fe@CeO2-Au/CN 椭球催化剂具有最高的反应速率常数k值和转化频率TOF值,从而表现较为优异的催化效率,该椭球催化剂独特的双空腔核壳结构特征和复合组分协同增强性效应是提升其催化反应性能的重要因素.
循环使用性能是催化剂另一个重要指标,影响到催化剂的使用寿命和使用成本. Fe@CeO2-Au/CN椭球催化剂使用完后经过磁分离回收,以及NaBH4溶液和去离子水先后淋洗再生处理后继续进行下一个循环使用.图9f显示,经过8次循环反应后,Fe@CeO2-Au/CN催化剂催化还原对硝基苯酚和有机染料的反应性能依然保持较高水平.图10a的TEM图显示多次使用后回收的Fe@CeO2-Au/CN椭球仍保留较完整的形貌结构,且图10b显示其保存大部分饱和磁化强度,表明该椭球催化剂具有极高的催化稳定性和循环使用性能.相反,图9f显示,Fe@CeO2-Au微粒在重复使用8次后,由于在回收过程中没有外部CN壳层的保护,Au颗粒脱落,其催化性能急剧下降.其原因在于固载的Au颗粒很容易从Fe@CeO2内核表面脱落,减弱了其协同作用,从而使催化活性急剧恶化.因此,外层CN壳结构可以作为空间屏障,有效抑制Au颗粒的团聚变形和脱离损失,显著提高催化剂的催化活性和稳定性.
基于上述结果,Fe@CeO2-Au/CN椭球催化剂的催化性能优越,易于回收和循环使用,具有较高的反应速率常数k值和转换频率TOF值,其催化还原反应机理如图11所示.
当Fe@CeO2-Au/CN椭球进入反应体系中,污染物分子和NaBH4通过CN介孔壳层扩散并到达Au活性位点表面,从而实现从给体BH4-到受体污染物分子的电子传递,然后迅速开启还原反应过程,其催化性能主要取决于负载Au颗粒的催化活性[19].Fe@CeO2-Au/CN椭球独特的双空腔核壳结构及其复合组分协同增强效应可以诱导该类还原反应形成高效的电子传递系统,其具体体现为:①CN介孔壳层具有固有疏水性和强π-π叠加作用,极大地增强了反应物的静电吸附能力,以及良好的电子导电性,极大促进电子从CN界面转移到Au位点,显著提高了催化性能[34];②可移动的Fe@CeO2内核中CeO2层可分散固载的纳米Au颗粒,以增加催化活性位点的暴露,并与之产生金属-载体化学效应,在Au-CeO2杂化界面上产生新的高活性位点[34];③CeO2层可以提高表面电子/电荷分离效率,其表面缺陷会进一步修饰纳米Au颗粒的电子结构,稳定其氧化态,产生强烈的界面效应,从而提高催化效率[35]. 乙二胺在这个过程中发挥稳定固载的纳米Au颗粒的作用,以及改善CN壳层碳结构导电性作用. 此外,由于Ce4+/Ce3+的标准氧化还原电位(1.44 V)高于Fe3+/Fe2+(0.77 V),CeO2和Fe在BH4-环境中的竞争氧化还原反应是热反应,有利于提高Fe2+向Ce4+的电子转移速率[36].Ce4+能够在Fe2+和BH4−存在的情况下进行Ce4+/Ce3+氧化还原循环,Fe3+也参与Fe3+/Fe2+氧化还原循环生成Fe2+,使衍生电子富集在Au位上[36].同时,足够多的BH4-离子进入Au位点,释放表面附着的H自由基[21].这些活性H自由基和电子通过Au位点迅速传递到吸附的污染物分子上,引发该污染物发生还原降解反应.
-
本文采用乙二胺介导的Stöber-SiO2/RF扩展法以及碳化-水热刻蚀得到Fe2O3@CeO2/CN核壳椭球,采用Au(en)2Cl3为前驱物的沉积沉淀反应-还原气氛热处理法原位构筑分散度较好的超细纳米Au粒子和小体积Fe内核颗粒并形成内空腔,得到Fe@CeO2-Au/CN双空腔核壳磁性椭球催化剂.乙二胺在Stöber-SiO2/RF反应中作为碱性催化剂和氮源,利用反应速率差异促使形成叠层复合物;其在纳米Au颗粒固载过程中起到稳定剂作用,在较高还原温度下促使形成纳米Au粒子的封装固化和强磁性能的同步原位整合.独特的双空腔核壳磁性结构特征及复合组分协同增强效应使该椭球催化剂表现出优异的催化反应活性和稳定性;同时,该催化剂具有显著的磁分离回收性能和循环使用性能,8次循环使用后仍保留较高的反应效率和结构完整性.CN介孔壳层的强π-π叠加作用和良好的电子导电性、金属-载体化学效应所产生的新的高活性位点、反应界面电子迁移速率的提高等方面是增强该催化剂催化性能的重要因素.
Fe@CeO2/CN双空腔核壳磁性载金椭球催化剂及其催化还原对硝基苯酚和染料污染物
Fe@CeO2/CN double-cavity core-shell magnetic Au-loaded ellipsoidal catalysts for catalytic reduction of 4-nitrophenol and dye pollutants
-
摘要: 载金催化剂可在温和条件下还原性降解环境中某些有毒有害污染物,并转化为低毒性、高附加值的物质,从而促进水体污染物化学资源转化和综合利用.本文设计制备Fe@CeO2/CN双空腔核壳磁性载金椭球催化剂,用于还原降解水体中对硝基苯酚和染料污染物.该催化剂先制备Fe2O3@CeO2梭型微粒内核,采用乙二胺介导的Stöber扩展法在其表面合成SiO2@RF复合物,经过碳化-水热蚀刻得到具有介孔碳氮壳层的Fe2O3@CeO2/CN椭球;采用[Au(en)2]3+为金前驱体的沉积沉淀-还原气氛热处理法在上述椭球中构筑较好分散度超细纳米Au颗粒,同时Fe2O3转化为小体积Fe颗粒并形成内空腔,得到Fe@CeO2-Au/CN双空腔核壳磁性椭球催化剂.该催化剂独特的结构特征和复合组分协同增强性效应使其在还原降解对硝基苯酚和染料污染物中均表现优越的催化反应性能和循环使用性能,重复多次使用后仍保存良好的磁分离性和较好的结构完整性.Abstract: Gold-loaded catalysts can degrade some toxic and harmful pollutants in the environment and convert them into substances with lower toxicity and higher added value under mild conditions, thus promoting the conversion and comprehensive utilization of water pollutant chemical resources. This paper provides a novel synthesis strategy to prepare Fe@CeO2/CN double-cavity core-shell magnetic ellipsoids with Au nanoparticles as catalysts for the reductive degradation of 4-nitrophenol and dye pollutants in the aquatic environment. Fe2O3@CeO2 shuttle-like particles are prepared as the core of the catalysts, and SiO2@RF composites are synthesized on the surface by the ethylenediamine-mediated Stöber extended method. Fe2O3@CeO2/CN ellipsoids with mesoporous carbon-nitrogen shells are obtained through carbonization and alkaline etching. Using [Au(en)2]3+ as gold precursor, ultrafine Au nanoparticles with good dispersion are constructed in the composite ellipsoids by means of deposition-precipitation and reducing atmosphere heat treatment. At the same time, Fe2O3 cores are converted into small Fe particles and inner cavities are formed, obtaining Fe@CeO2-Au/CN core-shell magnetic ellipsoidal catalysts with double cavities. The unique structural characteristics of the catalysts and the enhanced synergistic effect of the composite components enable these catalysts to possess excellent catalytic reaction performance and recycling capability in the reductive degradation of 4-nitrophenol and dye pollutants. After repeated use, the catalysts still preserve good magnetic separation ability and original structural integrity.
-

-
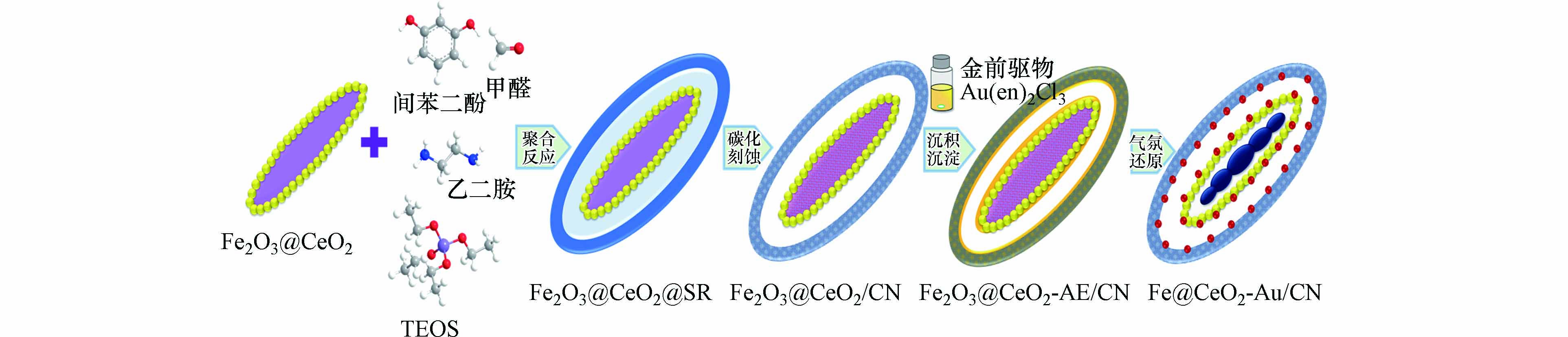
图 1 Fe@CeO2-Au/CN双空腔核壳磁性椭球催化剂的制备流程图
Figure 1. Preparation process of Fe@CeO2-Au/CN double-cavity core-shell magnetic ellipsoidal catalyst

图 2 Fe2O3@CeO2微粒(a),Fe2O3@CeO2@SR椭球(b),Fe2O3@CeO2/CN椭球(c)和Fe@CeO2-Au/CN椭球(d)的TEM图
Figure 2. TEM images of Fe2O3@CeO2 particles (a), Fe2O3@CeO2@SR ellipsoids (b), Fe2O3@CeO2-Au/CN ellipsoids (c) and Fe@CeO2-Au/CN ellipsoids (d)

图 3 Fe@CeO2-Au/CN椭球的HRTEM图(a)和SEAD图(b)
Figure 3. HRTEM (a) and SEAD (b) images of Fe@CeO2-Au/CN ellipsoids

图 4 Fe@CeO2-Au/CN椭球的SEM图(a)和EDS图(b)
Figure 4. SEM (a) and EDS (b) images of Fe@CeO2-Au/CN ellipsoids
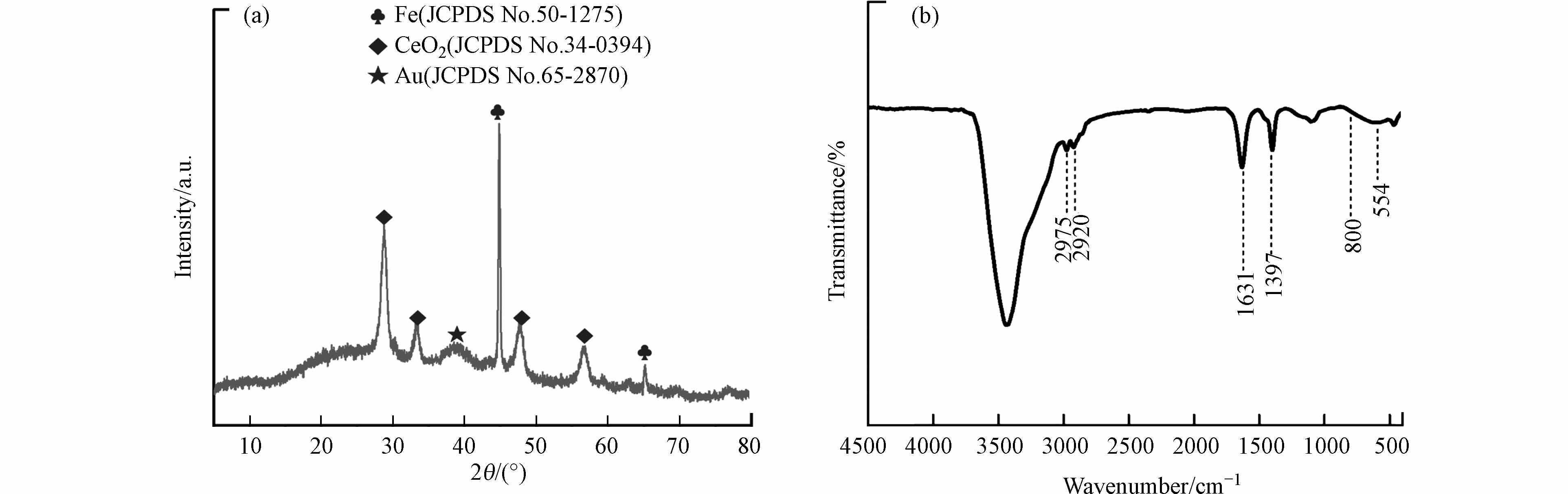
图 5 Fe@CeO2-Au/CN椭球的XRD图(a)和FTIR图(b)
Figure 5. XRD (a) and FTIR (b) patterns of Fe@CeO2-Au/CN ellipsoids

图 6 Fe@CeO2-Au/CN椭球的磁滞回线及磁回收性能图(a)和N2吸脱附曲线及孔径分布图(b)
Figure 6. The magnetization curve of Fe@CeO2-Au/CN ellipsoids and the picture of the magnetic separation (a); N2 absorption-desorption isotherm with pore size distribution curves for Fe@CeO2-Au/CN ellipsoids (b)

图 7 Fe@CeO2-Au/CN椭球的拉曼谱图(a)和UV-Vis漫反射图(b)
Figure 7. Raman (a) and UV-Vis DRS (b) spectra of Fe@CeO2-Au/CN ellipsoids

图 8 Fe@CeO2-Au/CN椭球的XPS谱图:(a) C 1s,(b) N 1s,(c) Ce 3d以及(d) Au 4f
Figure 8. XPS spectra of Fe@CeO2-Au/CN ellipsoids: (a) C 1s, (b) N 1s, (c) Ce 3d and (d) Au 4f
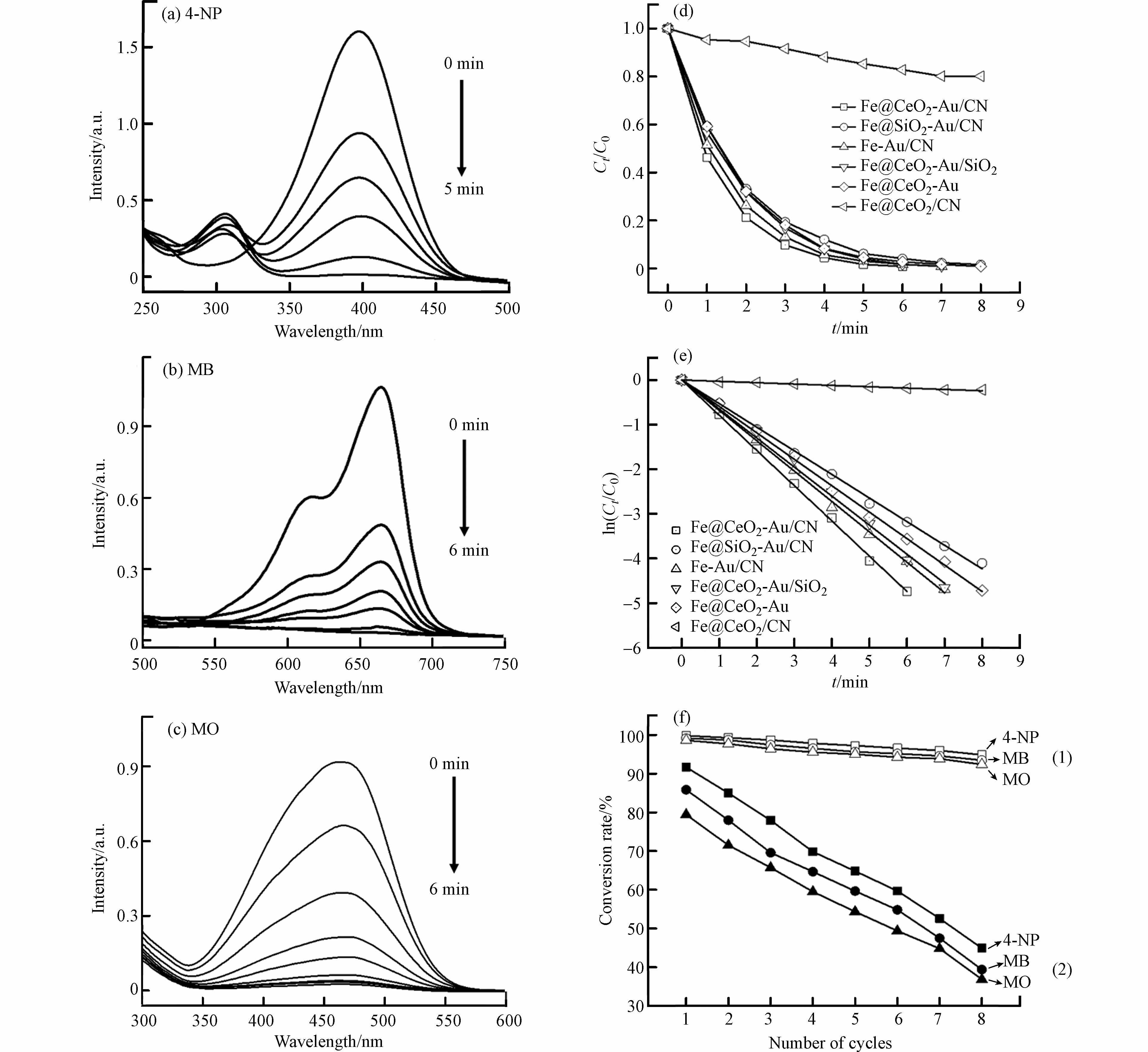
图 9 催化剂催化还原对硝基苯酚(a)、亚甲基蓝(b)和甲基橙(c)随时间变化的紫外可见光谱图;催化剂催化还原对硝基苯酚随时间变化的反应性能图(d)及一级反应动力学图(e);Fe@CeO2-Au/CN(1)和Fe@CeO2-Au(2)催化剂的循环反应性能图(f)
Figure 9. Time evolutions of UV-Vis absorption spectra for the reduction of 4-NP (a), MB (b) and MO (c) over Fe@CeO2-Au/CN catalyst; Plots of Ct/Co (d)and ln (Ct/Co)(e) versus reaction time for the reduction of 4-NP over different samples; Recyclability tests of Fe@CeO2-Au/CN (1)and Fe@CeO2-Au(2) catalysts (f)

图 10 多次催化反应后回收的Fe@CeO2-Au/CN椭球催化剂的TEM图(a)和磁化性能图(b)
Figure 10. TEM image (a) and magnetization curve (b) of the final recycled Fe@CeO2-Au/CN catalyst
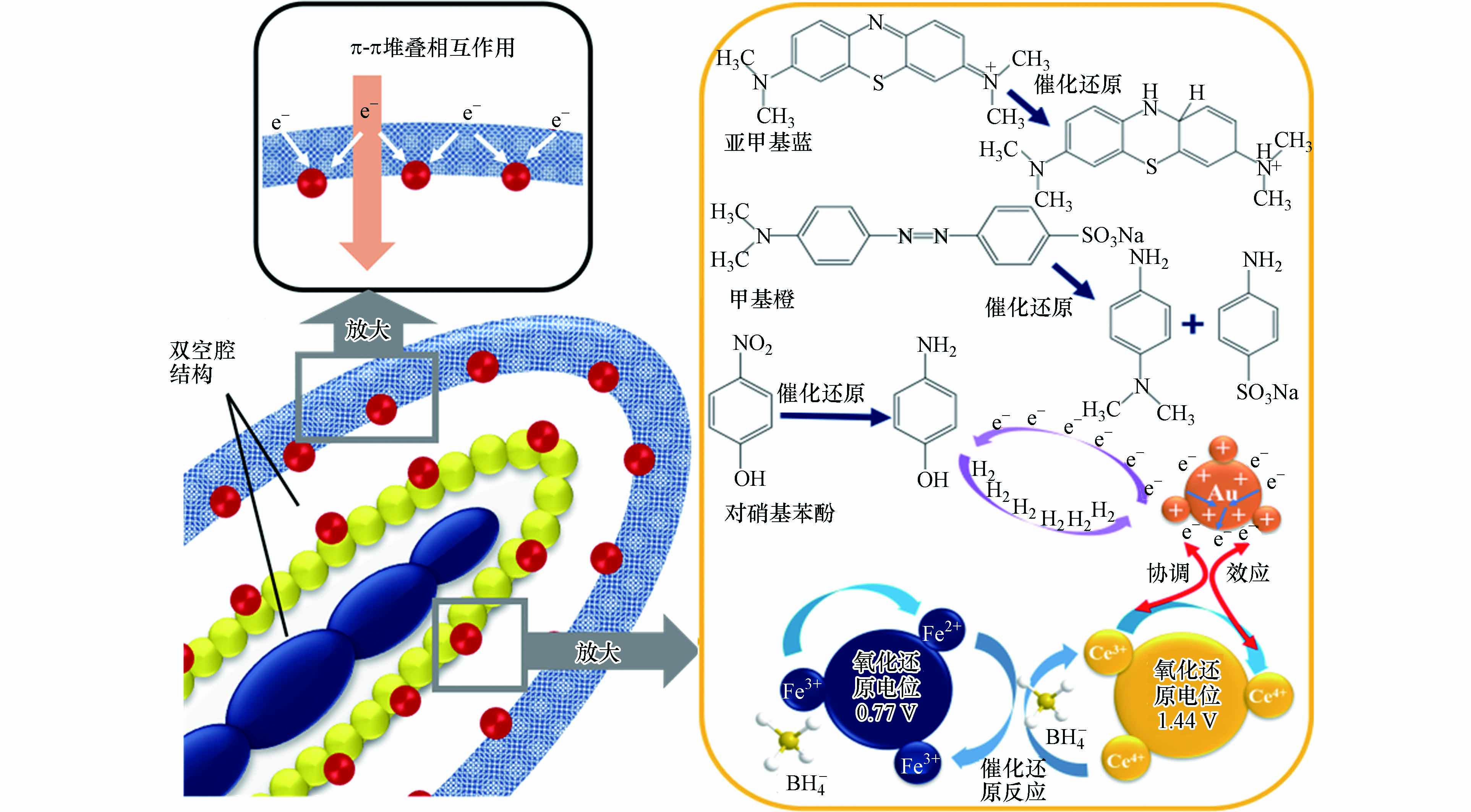
图 11 Fe@CeO2-Au/CN椭球催化剂的催化还原反应机理
Figure 11. Schematic of the reaction mechanism of Fe@CeO2-Au/CN ellipsoidal catalyst
表 1 Fe@CeO2-Au/CN催化剂与其他文献报道的载金催化剂性能对比
Table 1. Comparison of Fe@CeO2-Au/CN catalyst and other reported Au-supported catalysts in other literatures
催化剂
Catalyst反应时间/min
Reaction time反应速率常数/min−1
k Reaction rate constant转化频率/min−1
TOF Turnover frequency参考文献
ReferencesFe@CeO2-Au/CN椭球 6 0.790 16.83 本文 Fe@SiO2-Au/CN椭球 8 0.529 11.45 本文 Fe@Au/CN椭球 7 0.681 14.81 本文 Fe@CeO2-Au/SiO2椭球 7 0.650 6.47 本文 Fe@CeO2-Au微粒 8 0.592 9.29 本文 Fe@SiO2-Au/mSiO2微球 7.5 0.496 6.26 [21] Fe3O4@Au/mSiO2-TiO2微球 8 0.604 7.62 [24] Fe3O4@PDA-Au微球 6 0.630 4.10 [25] Au/CSNFs纳米纤维 16 0.354 9.38 [26] Au/SNTs纳米纤维 280 s 0.636 0.77 [27] Au/PAMAM空心微球 30 0.120 3.27 [28] Au/TiO2纳米纤维 16 0.244 0.97 [29] SiO2@Au/CeO2微球 5 0.780 4.0 [30] Fe3O4@Au/TiO2-ZrO2微球 6 0.369 3.56 [31] Au/CeO2空心微球 9 0.361 0.89 [32] H-TiO2/Au/H-mSiO2空心微球 9 0.303 1.25 [33]  下载: 导出CSV
下载: 导出CSV
-
[1] DAS R, SYPU V S, PAUMO K, et al. Silver decorated magnetic nanocomposite (Fe3O4@PPy-MAA/Ag) as highly active catalyst towards reduction of 4-nitrophenol and toxic organic dyes [J]. Applied Catalysis B:Environmental, 2019, 244: 546-588. doi: 10.1016/j.apcatb.2018.11.073 [2] CAI L Q, ZHANG L N, XU X J. One-step synthesis of ultra-small silver nanoparticles-loaded triple-helix β-glucan nanocomposite for highly catalytic hydrogenation of 4-nitrophenol and dyes [J]. Chemical Engineering Journal, 2022, 442: 136114. doi: 10.1016/j.cej.2022.136114 [3] 许婉馨, 杨波, 陈子伦, 等. 聚乙烯醇海绵负载铑催化剂催化还原对硝基苯酚 [J]. 环境化学, 2020, 39(9): 2576-2583. doi: 10.7524/j.issn.0254-6108.2020050803 XU W X, YANG B, CHEN Z L, et al. Polyvinyl sponge supported rh catalytic reduction of P-nitrophenol [J]. Environmental Chemistry, 2020, 39(9): 2576-2583(in Chinese). doi: 10.7524/j.issn.0254-6108.2020050803
[4] ASLAM S, SUBHAN F, YAN Z F, et al. Fabrication of gold nanoparticles within hierarchically ZSM-5-based micro/mesostructures (MMZ) with enhanced stability for catalytic reduction of pnitrophenol and methylene blue [J]. Separation and Purification Technology, 2021, 254: 117645. doi: 10.1016/j.seppur.2020.117645 [5] ZHU Q Y, ZHANG W J, CAI J, et al. Morphology-controlled synthesis of gold nanoparticles with chitosan for catalytic reduction of nitrophenol [J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2022, 640: 128471. doi: 10.1016/j.colsurfa.2022.128471 [6] CHEN C S, CHEN T C, CHIU T C, et al. Silver particles deposited onto magnetic carbon nanofibers as highly active catalysts for 4-nitrophenol reduction [J]. Applied Catalysis B:Environmental, 2022, 315: 121596. doi: 10.1016/j.apcatb.2022.121596 [7] DEVI A P, PADHI D K, MINHRA P M, et al. Bio-surfactant mediated synthesis of Au/g-C3N4 plasmonic hybrid nanocomposite for enhanced photocatalytic reduction of mono-nitrophenols [J]. Journal of Industrial and Engineering Chemistry, 2022, 108: 118-129. doi: 10.1016/j.jiec.2021.12.030 [8] NIU L, ZHAO X L, TANG Z, et al. Solid-solid synthesis of covalent organic framework as a support for growth of controllable ultrafine Au nanoparticles [J]. Science of The Total Environment, 2022, 835: 155423. doi: 10.1016/j.scitotenv.2022.155423 [9] WANG C, SONG F, WANG X L, et al. A cellulose nanocrystal templating approach to synthesize size-controlled gold nanoparticles with high catalytic activity [J]. International Journal of Biological Macromolecules, 2022, 209: 464-471. doi: 10.1016/j.ijbiomac.2022.04.046 [10] LIU Y L, DONG H L, HUANG H W, et al. Electron-deficient Au nanoparticles confined in organic molecular cages for catalytic reduction of 4-nitrophenol [J]. ACS Applied Nano Materials, 2022, 5(1): 1276-1283. doi: 10.1021/acsanm.1c03859 [11] CHEN H, ZHUANG Q, WANG H, et al. Ultrafine gold nanoparticles dispersed in conjugated microporous polymers with sulfhydryl functional groups to improve the reducing activity of 4-nitrophenol [J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2022, 649: 129459. doi: 10.1016/j.colsurfa.2022.129459 [12] LANDGE V, SONAWANE S, MANICKAM S, et al. Ultrasound-assisted wet-impregnation of Ag-Co nanoparticles on cellulose nanofibers: Enhanced catalytic hydrogenation of 4-nitrophenol [J]. Journal of Environmental Chemical Engineering, 2021, 9: 105719. doi: 10.1016/j.jece.2021.105719 [13] SHAH F, YADAV N, SINGH S. Phosphotungstate-sandwiched between cerium oxide and gold nanoparticles exhibit enhanced catalytic reduction of 4-nitrophenol and peroxidase enzyme-like activity [J]. Colloids and Surfaces B:Biointerfaces, 2021, 198: 111478. doi: 10.1016/j.colsurfb.2020.111478 [14] WU K, WANG X Y, Guo L L, et al. Facile synthesis of Au embedded CuOx-CeO2 core/shell nanospheres as highly reactive and sinter-resistant catalysts for catalytic hydrogenation of p-nitrophenol [J]. Nano Research, 2020, 12(8): 2044-2055. [15] MISRA M, CHOWDHURY S R, SINGH N. TiO2@Au@CoMn2O4 core-shell nanorods for photo-electrochemical and photocatalytic activity for decomposition of toxic organic compounds and photo reduction of Cr6+ ion [J]. Journal of Alloys and Compounds, 2020, 824: 153861. doi: 10.1016/j.jallcom.2020.153861 [16] GUO Z H, LU J S, XIE W S, et al. Facile preparation of recyclable Fe@metal phenolic networks-Au system for catalytic reduction of 4-nitrophenol [J]. Materials Chemistry and Physics, 2022, 281: 125907. doi: 10.1016/j.matchemphys.2022.125907 [17] Cao J R, Cai J H, Li R P, et al. A novel 3D yolk-double-shell Au@CdS/g-C3N4 nanostructure with enhanced photoelectrochemical and photocatalytic properties [J]. The Journal of Physical Chemistry C, 2022, 126: 4939-4947. [18] PENG H, WANG D, MA D S, et al. Multifunctional yolk-shell structured magnetic mesoporous polydopamine/carbon microspheres for photothermal therapy and heterogenous catalysis [J]. ACS Applied Materials & Interfaces, 2022, 14: 23888-23895. [19] FANG J S, ZHANG Y W, ZHOU Y M, et al. Synthesis of novel ultrasmall Au-loaded magnetic SiO2/carbon yolk-shell ellipsoids as highly reactive and recoverable nanocatalysts [J]. Carbon, 2017, 121: 602-611. doi: 10.1016/j.carbon.2017.06.022 [20] LIU C, WANG J, LI J S, et al. Controllable synthesis of N-doped hollowstructured mesoporous carbon spheres by anamine-induced Stöber-silica/carbon assembly process [J]. Journal of Materials Chemistry A, 2016, 4: 11916. doi: 10.1039/C6TA03748H [21] FANG J S, ZHANG Y W, ZHOU Y M, et al. Fabrication of ellipsoidal silica yolk-shell magnetic structures with extremely stable Au nanoparticles as highly reactive and recoverable Catalysts [J]. Langmuir, 2017, 33: 2698-2708. doi: 10.1021/acs.langmuir.6b03873 [22] GU S S, LOU Z, LI L D, et al. Fabrication of flexible reduced graphene oxide/Fe2O3 hollow nanospheres based on-chip micro-supercapacitors for integrated photodetecting applications [J]. Nano Research, 2016, 9(2): 424-434. doi: 10.1007/s12274-015-0923-7 [23] ZENG T, ZHANG X L, WANG S H, et al. A double-shelled yolk-like structure as an ideal magnetic support of tiny gold nanoparticles for nitrophenol reduction [J]. Journal of Materials Chemistry A, 2013, 1: 11641. doi: 10.1039/c3ta12660a [24] ZHANG H X, ZHANG Y W, ZHOU Y M, et al. Synthesis and characterization of a multifunctional nanocatalyst based on a novel type of binary-metal-oxide-coated Fe3O4-Au nanoparticle [J]. RSC Advances, 2016, 6: 18685. doi: 10.1039/C5RA27136C [25] ZENG T, ZHANG X L, NIU H Y, et al. In situ growth of gold nanoparticles onto polydopamine-encapsulated magnetic microspheres for catalytic reduction of nitrobenzene [J]. Applied Catalysis B:Environmental, 2013, 134-135: 26-33. doi: 10.1016/j.apcatb.2012.12.037 [26] KOGA H, TOKUNAGA E, HIDAKA M, et al. Topochemical synthesis and catalysis of metal nanoparticles exposed on crystalline cellulose nanofibers [J]. Chemical Communications, 2010, 46(45): 8567-8569. doi: 10.1039/c0cc02754e [27] HAN J, LI L Y, GUO R. Novel approach to controllable synthesis of gold nanoparticles supported on polyaniline nanofibers [J]. Macromolecules, 2010, 43(24): 10636-10644. doi: 10.1021/ma102251e [28] WU H Y, LIU Z L, WANG X D, et al. Preparation of hollow capsule-stabilized gold nanoparticles through the encapsulation of the dendrimer [J]. Journal of Colloid and Interface Science, 2006, 302(1): 142-148. doi: 10.1016/j.jcis.2006.06.019 [29] HAO Y G. , SHAO X K, LI B X, et al. Mesoporous TiO2 nanofibers with controllable Au loadings for catalytic reduction of 4-nitrophenol [J]. Materials Science in Semiconductor Processing, 2015, 40: 621-630. doi: 10.1016/j.mssp.2015.07.026 [30] LIU B C, YU S L, WANG Q, et al. Hollow mesoporous ceria nanoreactors with enhanced activity and stability for catalytic application [J]. Chemical Communications, 2013, 49: 3757-3759. doi: 10.1039/c3cc40665b [31] ZHANG H X, ZHANG Y W, ZHOU Y M, et al. Preparation of magnetically recoverable gold nanocatalysts with a highly reactive and enhanced thermal stability [J]. Journal of Alloys and Compounds, 2016, 688: 23-31. doi: 10.1016/j.jallcom.2016.07.019 [32] XU Y M, ZHANG Y W, ZHOU Y M, et al. CeO2 hollow nanospheres synthesized by a one pot template-free hydrothermal method and their application as catalyst support [J]. RSC Advances, 2015, 5: 58237-58245. doi: 10.1039/C5RA08124F [33] FANG J S, ZHANG Y W, ZHOU Y W, et al. In-situ construction of Au nanoparticles confined in double-shelled TiO2/mSiO2 hollow architecture for excellent catalytic activity and enhanced thermal stability [J]. Applied Surface Science, 2017, 392: 36-45. doi: 10.1016/j.apsusc.2016.08.157 [34] ZHANG J S, YAO T J, ZHANG H, et al. Preparation of raspberry-like γ-Fe2O3/crackled nitrogen-doped carbon capsules and their application as supports to improve catalytic activity [J]. Nanoscale, 2016, 8(44): 18693-18702. doi: 10.1039/C6NR05418H [35] EVANGELISTA V, ACOSTA B J, MIRIDONOV S, et al. Highly active Au-CeO2@ZrO2 yolk-shell nanoreactors for the reduction of 4-nitrophenol to 4-aminophenol [J]. Applied Catalysis B:Environmental, 2015, 166-167: 518-528. doi: 10.1016/j.apcatb.2014.12.006 [36] WANG Q, LI Y J, LIU B C, et al. Novel recyclable dual-heterostructured Fe3O4@CeO2/M (M= Pt, Pd and Pt-Pd) catalysts: synergetic and redox effects for superior catalytic performance [J]. Journal of Materials Chemistry A, 2015, 3: 139-147. doi: 10.1039/C4TA05691D -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1495
- HTML全文浏览数: 1495
- PDF下载数: 84
- 施引文献: 0
