

-
燃料高温燃烧过程中产生的氮氧化物(NOx)是大气的主要污染物之一,严重破坏生态环境并危害人类身体健康。还原或分解高温废气中的主要成分一氧化氮(NO)是控制NOx排放的主要方法。目前广泛应用的NO还原工艺以选择性非催化还原(Selective Non-Catalytic Reduction,SNCR)和选择性催化还原(Selective Catalytic Reduction, SCR)为主[1]。这些工艺中常用的还原剂氨具有高毒性和易燃性,同时SCR中催化剂的主要成分V2O5也有毒性,制约了这些工艺的经济性和安全性。因此,不用还原剂而将NO直接分解为N2和O2(2NO → N2 + O2)是去除NOx最理想的途径,但需要合适的催化剂来降低该反应的自由能[2-3]。
除了贵金属之外,直接分解NO的催化剂主要有钙钛矿型氧化物[4-5]、分子筛型催化剂[6-7]和其他复合氧化物[8-11]三类。由于C型立方结构的稀土氧化物(Rare earth oxide, REO)基催化剂[12-14]具有较高的NO转化率和较宽的工作温度窗口,逐步得到了广泛的关注和研究。在C型立方结构的稀土氧化物上,气相NO首先吸附在氧化物阴离子上并在表面形成硝酸盐物种(步骤一),这些物种在873 K以上的高温下转变为亚硝化基团(NO—),并与气相NO发生反应生成N2和O2(步骤二),实现NO的直接分解。其中,表面碱性中心是步骤1中NO吸附的关键,而氧化物阴离子空位是步骤2中亚硝基形成的关键[15]。Imanaka等[13]推测,C型立方结构中的氧空位增加了NO吸附概率的同时,稀土元素的氧化还原反应加速了表面氧的解吸速率,使得REO基催化剂表现出对NO的高分解活性。
REO基催化剂种类很多,现阶段的研发重点是在REO上负载碱土金属氧化物,如Ba、Sr、Ca、Mg等,增加氧空位的同时改善催化剂对NO的选择性吸附,提高NO的分解活性。研究结果表明,负载BaO表现出最高的NO分解活性,其次是SrO、CaO和MgO[16]。通过在不同REOs如Dy2O3、Sm2O3、Y2O3、La2O3、CeO2等中添加BaO,Doi等[10]发现BaO/Dy2O3表现出最高的NO分解效率,BaO/Sm2O3和BaO/Y2O3次之。由于Y2O3相对廉价且对NO的分解活性也很高,成为了最理想的REO载体之一。Ishihara等发现,Ba/BaY2O4[17]、Ba/Y2O3[11]和Ba3Y3.4Sc0.6O9[18]显示出相对较高的NO分解活性。Tatsumi等[19]发现,在1123 K条件下,Y2O3中添加BaO和Ni的催化剂对He气氛中0.5%体积NO的分解效率可以达到95%。此外,Tsujimoto等[20]发现,C型立方结构的Y2O3-ZrO2中添加BaO后,即使在O2、H2O或CO2的存在下也表现出较高的NO分解活性,有效地缓解了CO2吸附引起的催化剂中毒程度。
目前,直接分解NO方法的应用还具有一定的难度。首先,NO直接分解的有效工作温度通常需要1173 K以上,纳米级的催化剂颗粒容易烧结;其次,尽管REO基催化剂能削弱废气中的CO2、H2O和O2等气体对NO吸附位点的竞争,但NO分解活性仍然会大幅下降,进而导致NO分解效率降低;第三,大烟气量时所需的催化剂体积巨大,稀土的高成本制约着技术的推广应用。
针对以上问题,本研究尝试使用微米级C型立方晶体材料Y2O3作为基体材料,并通过混合一定比例的BaO和ZrO2获得混合型催化剂。在高温管式炉中考察了NO在混合型催化剂上的直接分解特性以及含氧气体CO、CO2和O2等对NO分解活性的影响,为经济耐高温型NO直接分解催化剂的制备和使用条件提供参考依据。
-
本研究中所用到的催化剂原料分别为:Y2O3(微米级,纯度≥99.99%),BaO(微米级,纯度≥97%),ZrO2(纳米级,纯度≥99%),通过机械混合获得一定质量配比的混合物。以质量比为0.233/1/9的ZrO2/BaO/Y2O3混合催化剂材料制备为例,分别称取Y2O3、BaO和ZrO2粉末7.74、0.86、0.2 g置于烧杯中,加入少量无水酒精覆盖材料后磁力搅拌10 min,随后在400 K的温度下烘干酒精获得混合均匀的催化剂。酒精湿混不仅加快了粉末的均匀混合,而且消除了催化剂粉末的飞扬。
-
采用荷兰PANalytical公司X’Pert Pro MPD型X射线衍射仪对催化剂的晶相进行分析,扫描范围为5°—90°,CuKα靶(入射光波长为0.154 nm),管电压40 kV,管电流30 mA,扫描速度10(°)·min−1,步长0.02(°)·s−1。采用BET(Brunauer-Emmett-Teller)法,用美国Quadrasob公司的SI-MP-10型比表面积分析仪在77 K下使用氮气吸附测量样品的比表面积。
-
采用石英玻璃管(内径10 mm)固定床流动反应器对NO进行了直接分解。首先在常温常压下称取2—2.2 g催化剂装填于反应器中部,并在两侧用石英棉固定。随后将用氩气Ar稀释的含NO的气体混合物送入催化剂床。气体流量由皂膜流量计标定后的质量流量计(MKS, GE50A系列)控制,实验中保持初始气体总流量为20 cm3·min−1。进料速率为W/F(g·s·cm−3),其中W和F分别为催化剂重量(g)和气体流量(cm3·s−1)。反应在473—1473 K的温度范围内进行。使用烟气分析仪(Testo 350)测定反应后气体中的NO、NO2和CO浓度,用气相色谱分析仪(Agilent 7890A)测定CO2和N2浓度。NO和CO的转化率根据入口和出口浓度之间的差异计算,公式如下:
式中,Y为NO或CO的转化率;C1和C2分别为相应气体的进口和出口体积分数(%)。由于CO和NO的体积分数很低,因此,这里忽略了反应引起的气体总量变化,由此引入的最大误差<3%。
-
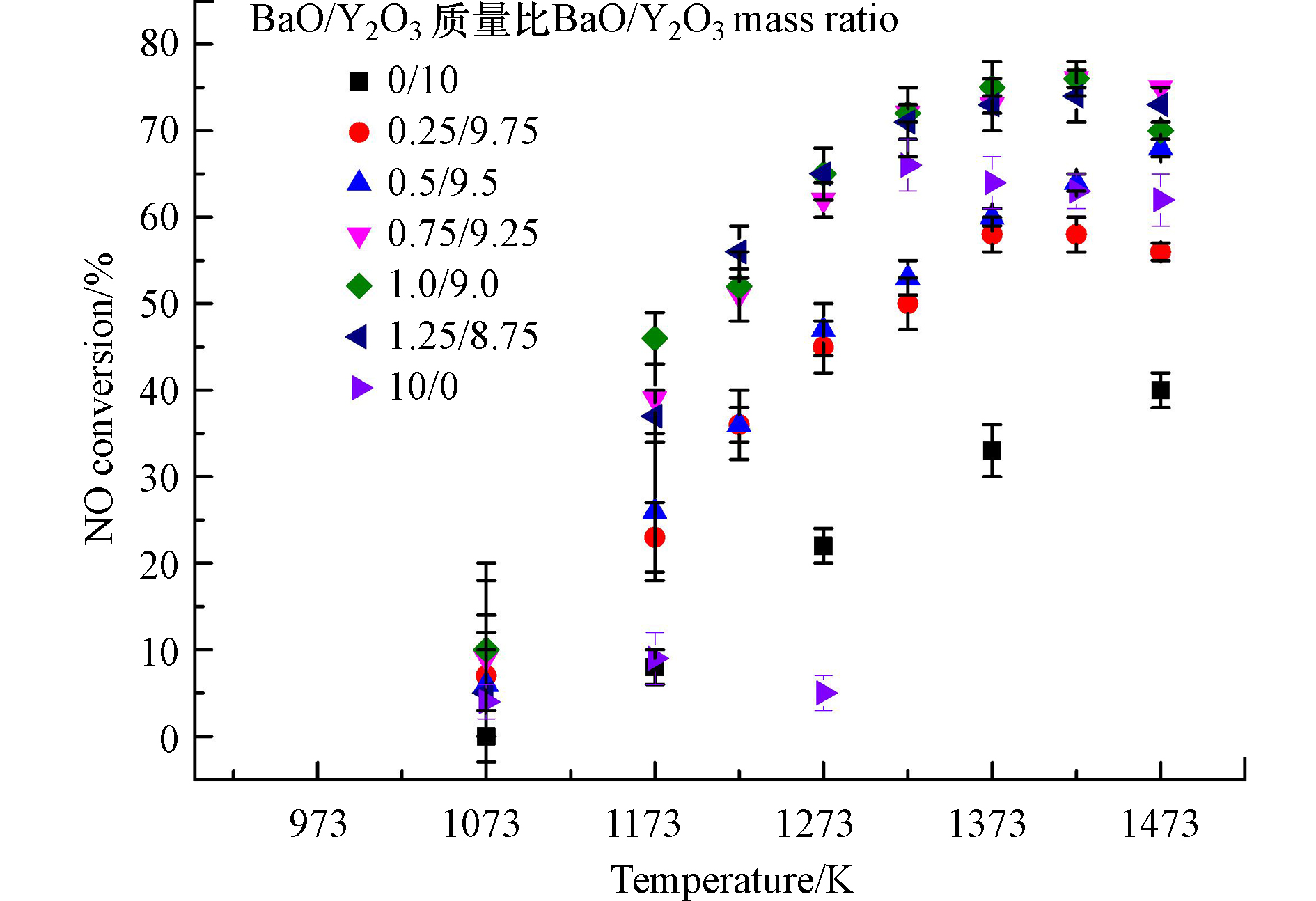
图1给出了不同BaO/Y2O3质量比的混合催化剂上NO转化率与反应温度的关系。由于混合催化剂对NO的分解活性在1073 K以上时才显现,因此只给出1073 K以上的实验结果。可以看出,纯Y2O3上NO的转化率随反应温度的升高而单调增加,在1473 K时达到40%。纯BaO的NO转化率在1273 K以下时低于10%,在1323 K时迅速跃升至66%,之后随温度升高略有下降。
添加少量BaO到Y2O3中后,混合催化剂的NO转化率有较大幅度提升。在1273 K以下时,混合催化剂的NO转化率远高于纯Y2O3,这是因为碱性的BaO可以大量吸附酸性的NO,尽管BaO在低温下对NO的分解活性很差,但混合催化剂中的Y2O3对吸附的NO有较高的分解活性,使得低温下的NO分解率升高。当温度高于1323 K时,NO转化率随着BaO添加比例的增加首先上升,并在BaO/Y2O3质量配比为1/9时达到最高,其在1423 K时的NO转化率可达76%。之后,继续增加BaO的比例使NO转化率略有下降,这是因为过多的BaO反而会减少Y2O3的暴露表面,降低混合催化剂整体的NO分解活性。由于烟气分析仪仅检测到极其微量的NO2,因此,可以近似认为NO在该催化剂上以化学计量方式分解转化成N2和O2。
-
在最佳配比为1/9的BaO/Y2O3中添加了少量的ZrO2以进一步提升混合催化剂高温分解NO的性能。图2给出了不同ZrO2/BaO/Y2O3质量比的混合催化剂上NO的转化率。由于纯ZrO2对NO没有分解活性,因此NO转化率为0。可以看出,尽管BaO/Y2O3上NO转化率在1373 K后开始下降,但是添加ZrO2后NO转化率随反应温度的升高单调增加,表明ZrO2改善了混合催化剂的高温催化活性。其次,NO转化率首先随着ZrO2比例的增加而升高,并在ZrO2/BaO/Y2O3的质量比为0.233/1/9时达到最高,在1473 K时NO转化率可达93%。之后,继续增加ZrO2的比例使NO转化率略有下降。Masaki等[12]认为,Zr4+替代Y3+可以有效提升NO直接分解活性,但是由于Zr4+离子半径较小,Zr4+离子比例过高会导致催化剂晶格中的空隙减少,进而导致NO吸附的减少和NO分解率的下降。因此,存在一个最优的ZrO2掺混比例以获得最佳的NO分解性能。
-
质量比为0.233/1/9的ZrO2/BaO/Y2O3混合催化剂上CO、CO2或O2的存在对NO转化率的影响如图3所示。当少量CO存在时,NO直接分解所需的温度大幅降低,而NO转化率大幅度上升,在1073 K时已经达到100%。同时,CO也全部转化为CO2。这是由于在催化剂的作用下CO可以与NO快速发生反应(2NO + 2CO = 2CO2 + N2),加速了NO的分解并最终生成N2和CO2。
当气氛中含有CO2时,由于CO2毒化作用,整个温度窗口ZrO2/BaO/Y2O3混合催化剂对NO的分解活性显著下降。这是因为酸性的CO2在催化剂表面具有强吸附作用,尤其是在碱土金属离子Ba2+位置上的吸附显著抑制了NO的吸附[21],而由图1可知,Ba2+是ZrO2/BaO/Y2O3催化剂直接分解NO的关键成分之一。如图3所示,在5%CO2存在情况下,NO转化率随温度从1073 K时的5%单调上升到1473 K时的73%,表明ZrO2/BaO/Y2O3混合催化剂仍具有一定的NO分解活性。
O2对催化剂上NO分解活性的抑制更为显著。一方面,O2的存在使得NO直接分解反应向N2生成方向的选择性降低,另一方面,NO与O2之间存在竞争吸附,大量O2占据了催化剂表面的活性位点,导致NO被吸附分解的概率大大降低。然而,图3显示,即使在5%氧气共存的情况下,NO转化率在1273 K时也有20%,而且提高温度能加速O2的脱附,使得1473 K时NO转化率能达到46%。类似于Y2O3–ZrO2固溶体[12],Y2O3晶格中掺杂ZrO2产生的Y3+阳离子空位或过量的氧化物阴离子有助于NO在氧共存下的高催化分解活性。
-
质量比为0.233/1/9的ZrO2/BaO/Y2O3混合催化剂直接分解NO的重复性实验结果如图4所示。单次实验中,催化剂经历从室温到1473 K的大幅度温度变化,持续时间约4 h。可以看到,经历多次重复实验后,催化剂的NO分解活性没有出现下降。同时,结合图3所示不同气氛条件下的多次实验结果表明,本研究获得的催化剂能够在1473 K的高温下长期稳定工作,并保持了对NO分解的高活性。
-
图5显示了不同比例ZrO2/BaO/Y2O3混合催化剂的XRD衍射图谱。在进行表征前,混合催化剂在1473 K的温度下煅烧1 h,以模拟流动反应器中的工作状态。对比纯Y2O3的衍射图谱可以发现,尽管在添加BaO和ZrO2后的衍射峰变宽,但在混合催化剂的衍射图谱中并未发现BaO与ZrO2的晶型结构,而主要是C型立方稀土氧化物结构的单相,伴随有BaCO3杂质的衍射峰。这可能是由于虽然催化剂制备为简单的物理混合,但在高温煅烧的过程中发生了固溶,实现了晶体掺杂[22],从而无法探测到BaO和ZrO2的衍射峰。由于六配位数的Y3+和Zr4+的离子半径分别为0.1040 nm和0.0860 nm[23],当Zr4+占据了晶格中Y3+的晶格位置,如图5所示,其衍射峰相对于Y2O3向更大角度移动,因此混合催化剂的晶格常数变小。通过XRD衍射图谱分析计算发现,质量比为1/9的BaO/Y2O3催化剂的晶格常数为10.5858,而添加ZrO2后质量比为0.233/1/9的ZrO2/BaO/Y2O3催化剂的晶格常数为10.5671,也表明C型立方固溶体的形成。
表1为不同质量比的ZrO2/BaO/Y2O3混合催化剂的BET比表面积。可以看出,BET比表面积随ZrO2比例的增加而单调增大,与图2中NO转化率之间没有直接关系。BET比表面积的增加一方面是Y2O3晶格中引入Zr4+导致,另一方面可能是由于纳米级ZrO2颗粒在微米级Y2O3颗粒表面发生固溶导致的表面形貌变化。与Tsujimoto[20]通过共沉淀法获得的Y2O3-BaO-ZrO2复合催化剂相比,本研究用物理混合法制备的ZrO2/BaO/Y2O3混合催化剂的BET比表面积仅为其1/20,这可能是在相同的1173 K条件下,本研究的NO转化率与Tsujimoto[20]的结果相比相差一倍的主要原因之一。因此,提高催化剂的比表面积是今后研究的主要工作之一。
-
对Y2O3、BaO和ZrO2的3种催化剂原料直接混合后高温煅烧,可发生固溶并形成C型立方结构的ZrO2/BaO/Y2O3混合催化剂。质量比为0.233/1/9的ZrO2/BaO/Y2O3混合催化剂具有最高的NO分解活性,可以在1473 K的高温下稳定工作且NO的转化率可以高达93%。值得注意的是,CO的协同效应可以使NO在更低的温度下完全分解,而O2或CO2的存在则会抑制NO的催化分解,但在高温下依然能保持较高的转化率。与结构精细设计的催化剂不同,本研究验证了混合煅烧有可能是一种经济可靠的高温下NO直接分解催化剂的制备途径。尽管本论文研究了不同气体对NO直接催化分解的影响,但真实烟气复杂气氛下的实验研究还需要进一步开展。
混合型催化剂BaO/ZrO2/Y2O3高温直接分解NO性能
Performance of NO direct decomposition over mixed catalyst BaO/ZrO2/Y2O3 at high temperature
-
摘要: 为改善更高温度下稀土氧化物Y2O3直接分解NO的性能,在管式炉中实验考察了混合BaO和ZrO2两种金属氧化物对NO分解效率的影响。结果显示,混合BaO和ZrO2可以改善Y2O3直接分解NO的活性,其中质量比为0.233/1/9的ZrO2/BaO/Y2O3混合催化剂的效果最好,在1473 K、NO/Ar气氛下NO分解效率可达93%。当气氛中存在CO2和O2时,尽管温度窗口内催化剂直接分解NO的活性降低,但1473 K时的分解效率仍可以维持45%以上。气氛中CO的共存能降低催化剂活化温度并协同实现NO完全分解。X射线衍射测试表明ZrO2/BaO/Y2O3在高温下发生固溶,Zr4+的掺杂使Y2O3晶格常数变小,生成了更适合NO吸附和分解的活性氧空位。本研究表明多种金属氧化物机械混合后高温煅烧是一种有前景的NO直接分解催化剂的制备方法。Abstract: In order to improve the NO direct decomposition of rare earth oxide Y2O3 under high temperatures, the effects of BaO and ZrO2 mixing on the NO conversion efficiency were investigated in a tubular furnace. The results show that mixing Y2O3 with BaO and ZrO2 can increase the NO direct decomposition activity and the best mass ratio of ZrO2/BaO/Y2O3 is 0.233/1/9, which can show a high NO conversion efficiency of 93% at 1473 K under NO/Ar atmosphere. In the presence of CO2 and O2, although the activity of NO decomposition decreases in the temperature window, the NO conversion efficiency can still maintain above 45% at 1473 K. The coexistence of CO can lower the activation temperature of the catalyst and synergistically promote the complete decomposition of NO. The X-ray diffraction results show the formation of ZrO2/BaO/Y2O3 solid solution at high temperature, in which Zr4+ doping makes the lattice constant of Y2O3 smaller, resulting in active oxygen vacancies more suitable for NO adsorption and decomposition. These results suggest that mechanical mixing of various metal oxides followed by high temperature calcination is a promising method to prepare catalysts for NO direct decomposition.
-
Key words:
- rare earth oxide /
- yttrium oxide /
- NO /
- direct decomposition
-

-

图 2 ZrO2/BaO/Y2O3混合催化剂上NO的转化率
Figure 2. NO conversions over the ZrO2/BaO/Y2O3 mixed catalyst
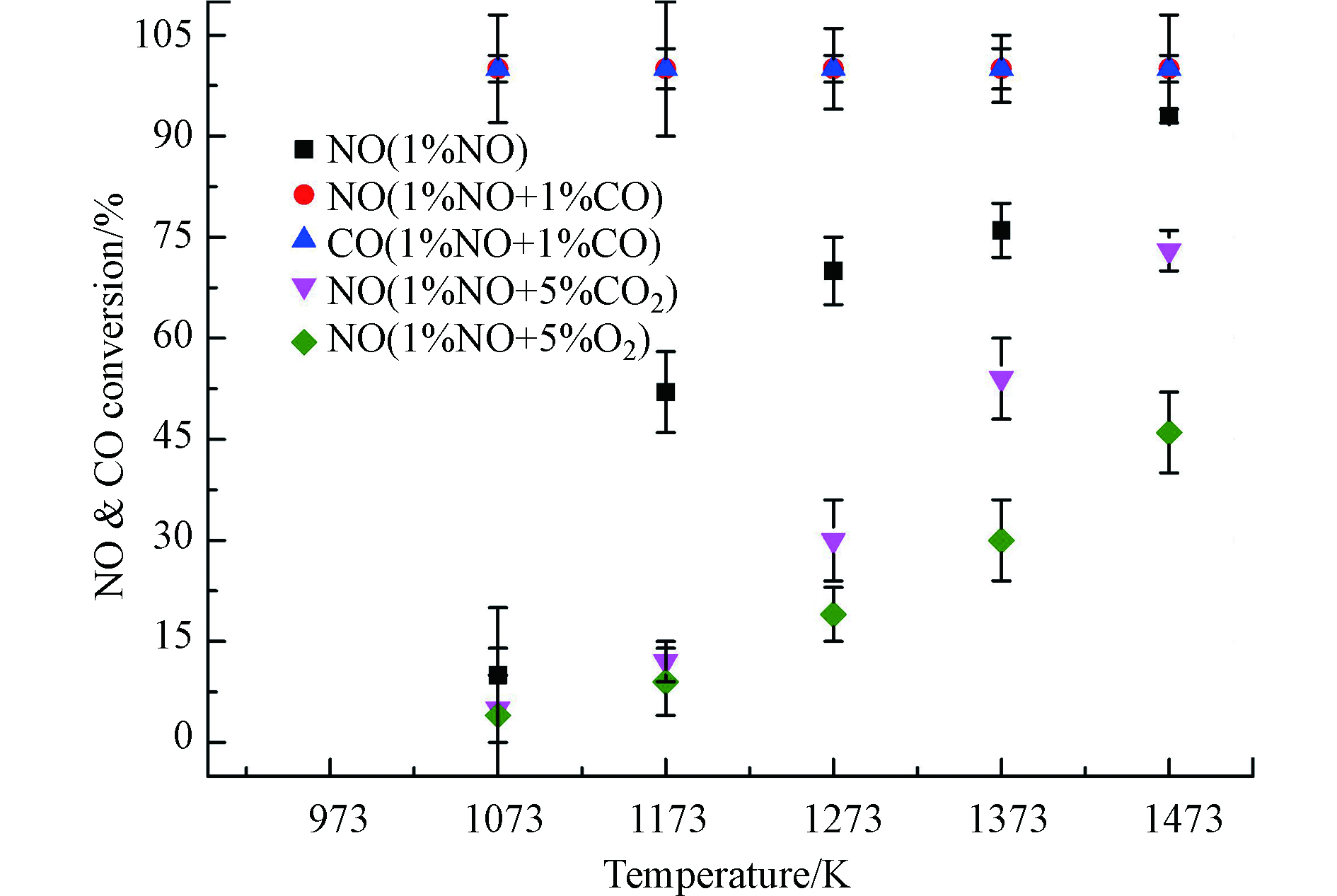
图 3 反应气氛中CO、CO2和O2对质量比为0.233/1/9的ZrO2/BaO/Y2O3混合催化剂上NO和CO转化率的影响
Figure 3. NO and CO conversions over the ZrO2/BaO/Y2O3 mixed catalyst with the mass ratio of 0.233/1/9 in the presence of CO, CO2 and O2
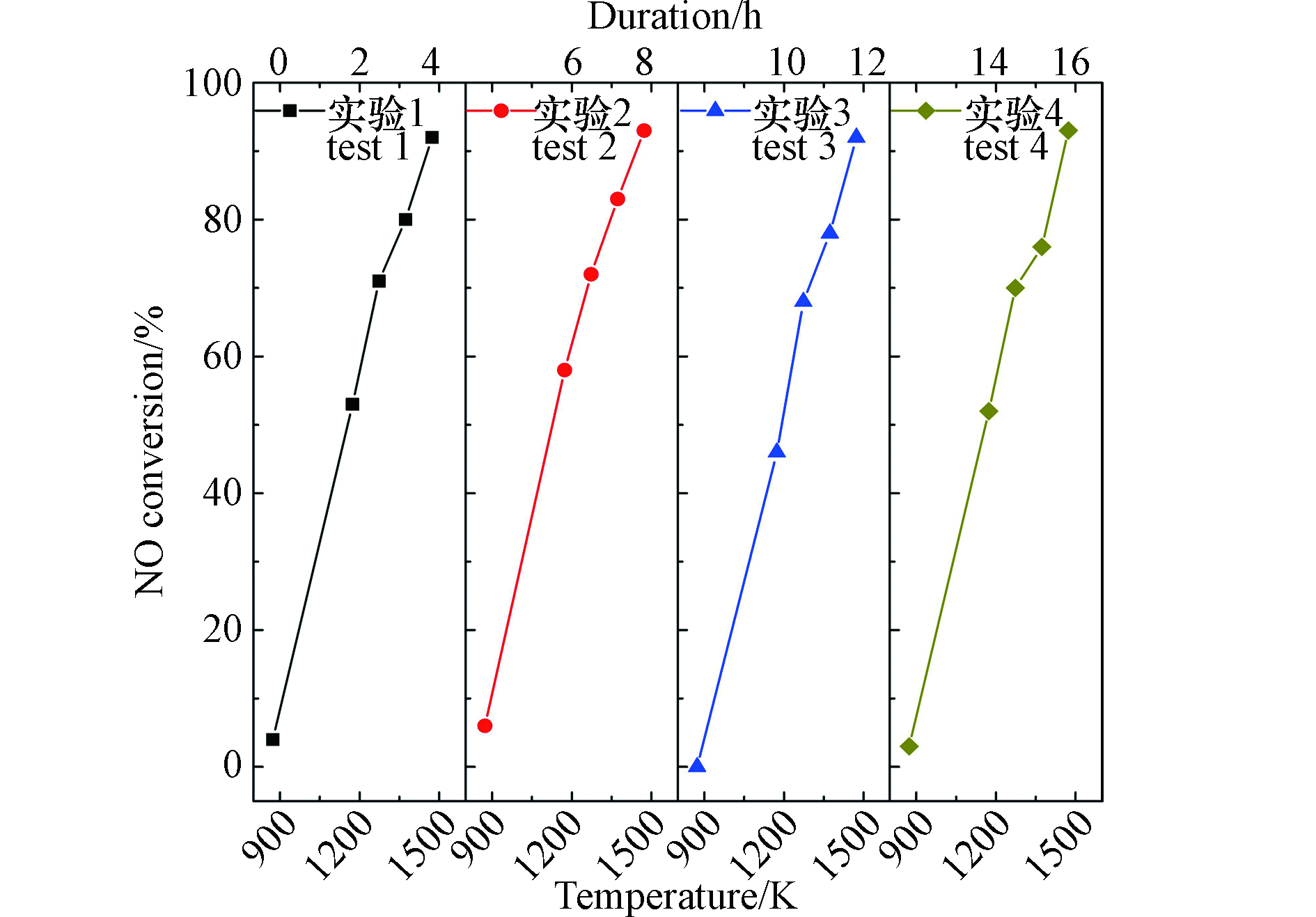
图 4 质量比为0.233/1/9的ZrO2/BaO/Y2O3混合催化剂NO分解活性的重复性实验
Figure 4. Repeatability test of NO decomposition activity over the ZrO2/BaO/Y2O3 mixed catalyst with the mass ratio of 0.233/1/9
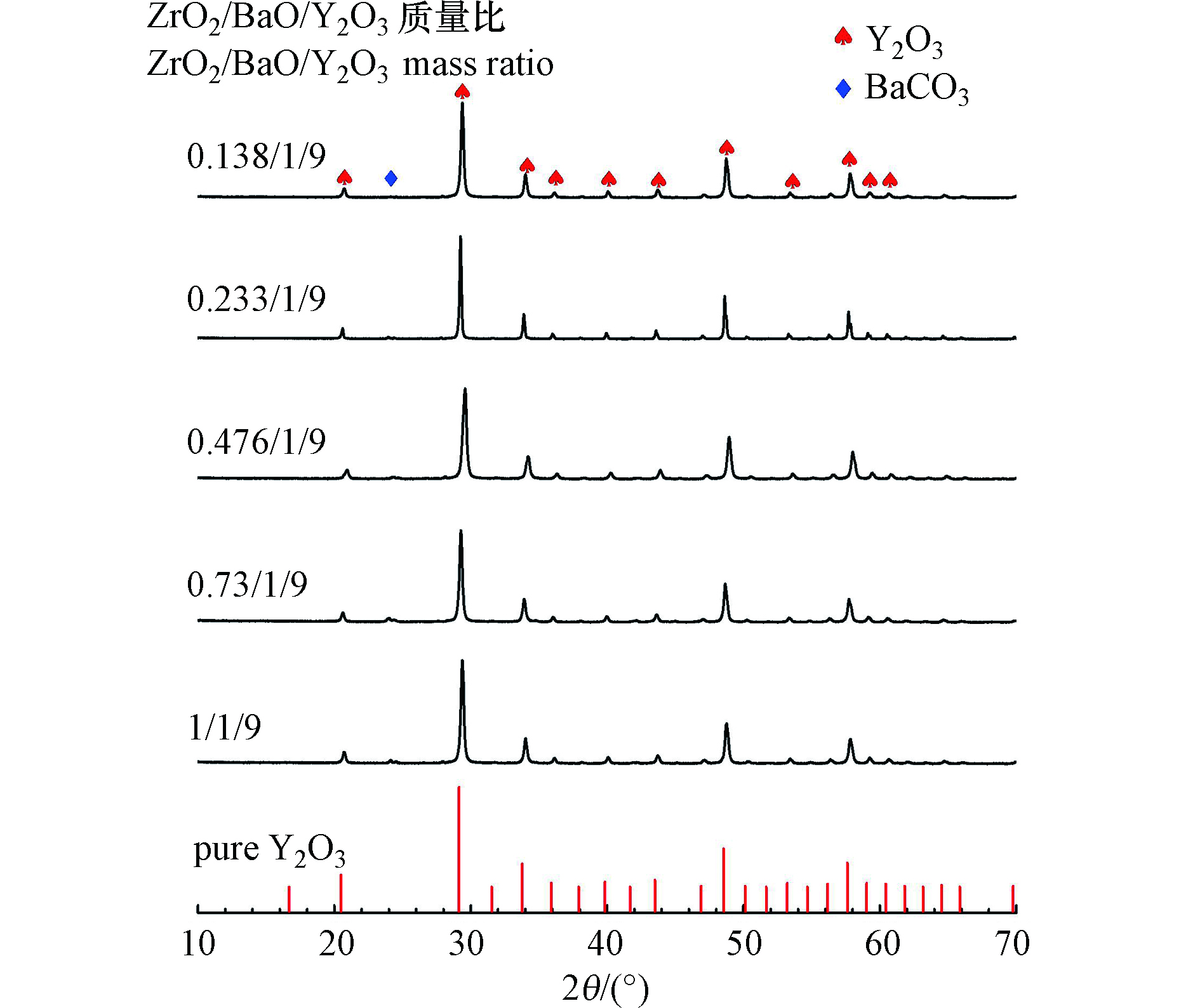
图 5 ZrO2/BaO/Y2O3混合催化剂的XRD衍射图谱
Figure 5. XRD patterns of the ZrO2/BaO/Y2O3 mixed catalysts.
表 1 ZrO2/BaO/Y2O3混合催化剂的BET比表面积
Table 1. BET surface area of the ZrO2/BaO/Y2O3 mixed catalysts
样品质量比
Mass ratio of samplesBET比表面积/(m2·g−1)
BET surface area0/1/9 1.626 0.138/1/9 1.630 0.233/1/9 1.641 0.476/1/9 1.737 0.73/1/9 1.964 1/1/9 2.370  下载: 导出CSV
下载: 导出CSV
-
[1] LIU Z M, IHL WOO S. Recent advances in catalytic DeNOX Science and technology [J]. Catalysis Reviews, 2006, 48(1): 43-89. doi: 10.1080/01614940500439891 [2] HANEDA M, HAMADA H. Recent progress in catalytic NO decomposition [J]. Comptes Rendus Chimie, 2016, 19(10): 1254-1265. doi: 10.1016/j.crci.2015.07.016 [3] SUN Q, WANG Z, WANG D, et al. A review on the catalytic decomposition of NO to N2 and O2: Catalysts and processes [J]. Catalysis Science & Technology, 2018, 8(18): 4563-4575. [4] ZHU J J, LI H L, ZHONG L Y, et al. Perovskite oxides: Preparation, characterizations, and applications in heterogeneous catalysis [J]. ACS Catalysis, 2014, 4(9): 2917-2940. doi: 10.1039/C8CY01114A [5] 马爱静, 王绍增, 邹鸿鹄, 等. La0.7Sr0.3Co1-xFexO3催化剂氮氧化物储存及抗硫性能 [J]. 物理化学学报, 2012, 28(6): 1474-1480. doi: 10.3866/PKU.WHXB201203311 MA A J, WANG S Z, ZOU H H, et al. The performance of the NOx storage capacity and sulfur tolerance of the La0.7Sr0.3Co1-xFexO3 catalyst [J]. Acta Physico-Chimica Sinica, 2012, 28(6): 1474-1480(in Chinese). doi: 10.3866/PKU.WHXB201203311
[6] 姜慧超, 赵朝成, 王一迪. 直接法合成杂原子Cu-ZSM-5用于直接催化分解NOx的研究 [J]. 现代化工, 2014, 34(11): 83-86. doi: 10.16606/j.cnki.issn0253-4320.2014.11.004 JIANG H C, ZHAO C C, WANG Y D. Direct synthesis of hetero atoms Cu- ZSM- 5 for direct catalytic decomposition of NOx [J]. Modern Chemical Industry, 2014, 34(11): 83-86(in Chinese). doi: 10.16606/j.cnki.issn0253-4320.2014.11.004
[7] 陈艳平, 程党国, 陈丰秋, 等. Cu-ZSM-5分子筛催化分解及选择性催化还原NO[J]. 化学进展, 2014, 26(增刊1): 248-258. CHEN Y P, CHENG D G, CHEN F Q, et al. NO decomposition and selective catalytic reduction of NO over Cu-ZSM-5 zeolite[J]. Progress in Chemistry, 2014, 26(Sup 1): 248-258(in Chinese).
[8] IMANAKA N, MASUI T. Advanced materials for environmental catalysts [J]. Chemical Record, 2009, 9(1): 40-50. [9] BION N, CAN F, COURTOIS X, et al. Transition metal oxides for combustion and depollution processes[M]//Metal Oxides in Heterogeneous Catalysis. Amsterdam: Elsevier, 2018: 287-353. [10] DOI Y, HANEDA M, OZAWA M. Direct decomposition of NO on Ba catalysts supported on rare earth oxides [J]. Journal of Molecular Catalysis A:Chemical, 2014, 383/384: 70-76. doi: 10.1016/j.molcata.2013.11.033 [11] ISHIHARA T, GOTO K. Direct decomposition of NO over BaO/Y2O3 catalyst [J]. Catalysis Today, 2011, 164(1): 484-488. doi: 10.1016/j.cattod.2010.12.005 [12] MASAKI H, MASUI T, IMANAKA N. Direct decomposition of nitric oxide into nitrogen and oxygen over C-type cubic Y2O3-ZrO2 solid solutions [J]. Journal of Alloys and Compounds, 2008, 451(1/2): 406-409. [13] IMANAKA N, MASUI T. Advances in direct NOx decomposition catalysts [J]. Applied Catalysis A:General, 2012, 431/432: 1-8. doi: 10.1016/j.jallcom.2007.04.158 [14] MASUI T, UEJIMA S, TSUJIMOTO S, et al. Direct NO decomposition over C-type cubic Y2O3-Pr6O11-Eu2O3 solid solutions [J]. Catalysis Today, 2015, 242: 338-342. doi: 10.1016/j.cattod.2014.05.047 [15] TSUJIMOTO S, YASUDA K, MASUI T, et al. Effects of Tb and Ba introduction on the reaction mechanism of direct NO decomposition over C-type cubic rare earth oxides based on Y2O3 [J]. Catalysis Science & Technology, 2013, 3(8): 1928-1936. doi: 10.1039/C3CY20746C [16] TSUBOI G, HANEDA M, NAGAO Y, et al. Direct decomposition of NO over supported-alkaline earth metal oxide catalysts [J]. Journal of the Japan Petroleum Institute, 2005, 48(1): 53-59. doi: 10.1627/jpi.48.53 [17] GOTO K, MATSUMOTO H, ISHIHARA T. Direct decomposition of NO on Ba/Ba-Y-O catalyst[J]. Topics in Catalysis, 2009, 52(13/14/15/16/17/18/19/20): 1776-1780. [18] GOTO K, ISHIHARA T. Direct decomposition of NO into N2 and O2 over Ba3Y3.4Sc0.6O9 [J]. Applied Catalysis A:General, 2011, 409/410: 66-73. doi: 10.1007/s11244-009-9337-7 [19] ISHIHARA T, FANG S M, IDE T. Effects of strain induced by Au dispersion in Ba and Ni doped Y2O3 on direct decomposition of NO [J]. Molecular Catalysis, 2019, 475: 110488. doi: 10.1016/j.mcat.2019.110488 [20] TSUJIMOTO S, WANG X J, MASUI T, et al. Direct decomposition of NO into N2 and O2 on C-type cubic Y2O3–ZrO2 and Y2O3–ZrO2–BaO [J]. Bulletin of the Chemical Society of Japan, 2011, 84(7): 807-811. doi: 10.1246/bcsj.20100360 [21] TSUJIMOTO S, NISHIMURA C, MASUI T, et al. Coexisting gas-resistant C-type cubic Yb2O3–Tb4O7 Catalysts for direct NO decomposition [J]. Chemistry Letters, 2011, 40(7): 708-710. doi: 10.1246/cl.2011.708 [22] de LEEUW D M, MUTSAERS C A H A, LANGEREIS C, et al. Compounds and phase compatibilities in the system Y2O3-BaO-CuO at 950℃ [J]. Physica C:Superconductivity, 1988, 152(1): 39-49. doi: 10.1016/0921-4534(88)90071-8 [23] SHANNON R D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides [J]. Acta Crystallographica Section A, 1976, 32(5): 751-767. doi: 10.1107/S0567739476001551 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 2700
- HTML全文浏览数: 2700
- PDF下载数: 93
- 施引文献: 0
