

-
重金属污染已经成为危害最大、范围最广的水污染物之一,是当今人类社会面临的一个严重问题。重金属具有毒性,且不可降解,除了对生活在水和土壤中的生物有毒害作用外,还会在整个食物链中积累,对人类、植物和动物的健康造成严重的危害。此外,重金属本身也是不可再生资源,因此,去除并可持续利用这些重金属的需求较为迫切,具有选择性地去除重金属离子方法的需求日益增加,其中常用的包括化学沉淀法[1]、离子交换法[2]、重金属捕集剂法[3]和吸附法[4]等。吸附法具有操作简便、成本低、效率高的特点而引起了广泛关注,而选择性吸附的关键在于吸附剂的选择与制备。
聚乙烯(PVA)((C2H4O)n)是聚醋酸乙烯酯的衍生物,是一种无毒、可降解、生物相容性好、成本相对较低的聚合物[5],因为聚乙烯醇中含有大量的羟基,可与多种官能团发生反应[6],可通过接枝对目标离子具有强作用力的官能团实现选择性吸附。细菌纤维素(BC)是一种新型的多功能纳米生物材料,是醋酸杆菌生长和代谢过程中分泌的高分子多糖[7],具有成本低、结构简单、亲水性好等特点,其主链上有大量的羟基,掺入PVA内可以有效提升PVA的力学性能[8]。硫脲中氨基官能团上的氢原子可被其他官能团取代生成硫脲基的衍生物,如脒基硫脲等。由于该类化合物上含有丰富的氮、硫元素,易与重金属离子通过配位作用生成稳定的螯合产物[9],可通过交联或接枝法负载到基质上,得到低成本、高选择性的吸附剂。
本研究以经戊二醛交联后的PVA/BC为基材,与脒基硫脲按照比例共混,采用水热法将脒基硫脲接枝到SPVA/BC表面,制备脒基硫脲改性交联聚乙烯醇/细菌纤维素的新型吸附剂(GLA-SPVA/BC)。通过FT-IR、SEM/EDS-mapping等表征手段对材料的形貌组成进行了表征并分析;通过吸附实验对其吸附热力学、动力学和选择性进行研究;对吸附动力学和热力学数据进行了拟合,与表征结果结合预测材料的吸附机理。
-
聚乙烯醇-124(PVA)、脒基硫脲(GLA)、硝酸锌:分析纯,这些分析纯试剂均来自国药集团化学试剂有限公司。戊二醛、硝酸铜、硝酸镍、硝酸铅、硝酸、盐酸:均为分析纯,购自天津科密欧化学试剂有限公司。细菌纤维素(BC)乳液:实验室自制。
-
称取0.7255 gPVA溶于20 mL去离子水中,并按照VPVA:VBC=4:1的比例加入BC乳液,该混合体系在90 ℃的恒温水浴锅中加热搅拌直至完全溶解并进行超声脱泡处理,配置成3.5%(质量百分比)的PVA/BC溶液。将该溶液缓慢的逐滴滴入pH=1.0(用浓盐酸调节)的30%的戊二醛溶液中,在30 ℃下搅拌2 h。待其交联完成,将最终得到的白色蓬松的固体研磨为粉末,用去离子水反复冲洗直至滤液呈中性,抽滤,烘干,并记为SPVA/BC。
称取脒基硫脲0.374 g并加入20 mL醋酸-醋酸钠缓冲溶液中,调节缓冲液为pH 4.6。待脒基硫脲完全溶解,向该混合体系中加入0.10 g材料SPVA/BC,在50 ℃的恒温水浴锅中搅拌反应3 h。将所得到的产物用去离子水反复冲洗直至滤液pH呈中性,抽滤,烘干,所得最终产品记为GLA-SPVA/BC。
-
FT-IR表征:使用Nicolet iS5红外光谱仪(美国ThermoFisher)中在400—4000 cm−1范围内检测红外吸收信号。SEM表征:使用Sigma HD扫描电子显微镜(德国Zeiss)在不同倍数下观察样品表面的微观形貌特征。TG-DSC表征:使用STA449F3同步热分析仪(德国NETZSCH)对样品材料GLA-SPVA/BC在30—600 ℃温度范围内进行加热并记录升温数据。XPS表征:使用Thermo ESCALAB 250XI光电子能谱仪对材料吸附前后原子特征结合能峰位变化进行检测。
-
分别称取5份质量为0.05 g的吸附剂SPVA/BC和GLA-SPVA/BC置于100 mL锥形瓶中,并加入含铜浓度为100.0 mg·L−1的Cu(NO3)2溶液50 mL,将锥形瓶置于120 r·min−1的恒温振荡箱(SHA-B,常州国华电器有限公司)振荡反应12 h,之后对溶液进行过滤定容,通过TAS-990原子吸收分光光度计(北京普析通用仪器有限公司)测定稀释后滤液中剩余的Cu(Ⅱ)浓度,通过计算,考察不同pH值对Cu(Ⅱ)吸附容量的影响。用1.0 mol·L−1硝酸调节溶液pH可进行初始pH影响的测定;调节恒温振荡箱的温度(20 ℃、30 ℃、40 ℃)并在不同时间点依次取样进行吸附动力学的测定;分别加入不同初始Cu(Ⅱ)浓度并调节恒温振荡箱的温度进行吸附热力学的测定;在四元混合溶液(均为100.0 mg·L−1)进行吸附选择性的测定。计算公式如下:
式中,qe为平衡吸附量,mg·g−1;C0、Ce为分别为Cu(Ⅱ)溶液的初始质量浓度和平衡质量浓度,mg·L−1;V为吸附液总体积,L;m为吸附剂质量,g。
-
图1为PVA、SPVA/BC和GLA-SPVA/BC复合材料的红外吸收光谱图,在3374 cm−1处为PVA终端—OH的伸缩振动[10];BC的特征吸收峰一般在3448 cm−1处[11],但由于加入的BC乳液量极少且BC的特征吸收峰与—OH的峰有所重叠,所以红外光谱图中未出现明显的分峰现象。PVA中1096 cm−1处的峰为长链中的—CO—基团的振动峰,戊二醛交联后,该峰在SPVA/BC和GLA-SPVA/BC中发生移动至1138 cm−1。此外,吸附剂GLA-SPVA/BC存在两个特征吸收峰,1514 cm−1处为C=N的伸缩振动峰,这是戊二醛的醛基和脒基硫脲中氨基交联产生的新键,874 cm−1处的键为脒基硫脲中的—(C=S)—N基团。这些特征吸收峰都可以表明脒基硫脲成功的接枝在SPVA/BC表面,脒基硫脲中的—(C=S)—N与Cu(Ⅱ)良好的配位作用为Cu(Ⅱ)的吸附提供了可能。
吸附剂GLA-SPVA/BC的拉曼光谱图如图1(b)所示。在662 cm−1为C—N的变形振动,表明吸附剂中有脒基硫脲的存在。在880 cm−1、992 cm−1和1143 cm−1处的特征谱带可能是长的碳氢链中C—C的伸缩振动或者是醚的对称/反对称振动,这是由于位于1200—800 cm−1范围内C—O的伸缩振动谱带与其他骨架振动重叠,这表明PVA与戊二醛发生了交联。此外,在1438 cm−1处的特征谱带为—NH3变形振动产生的谱峰,—NH3在2925 cm−1为合频产生的峰。
-
材料SPVA/BC (a)、GLA-SPVA/BC (b)和GLA-SPVA/BC-Cu (Ⅱ) (c)的SEM图像如图2所示。图2(a)和(b)中,SPVA/BC材料表面呈疏松大孔的状态;接枝脒基硫脲后,GLA-SPVA/BC材料相比SPVA/BC表面,空隙变小且变得粗糙。此外,图2b1为吸附剂GLA-SPVA/BC的EDS图像,图中明显可见S和N元素的谱带,并且图2b2、图2b3分别为S和N的mapping图像,S和N元素分布均匀,这表明脒基硫脲均匀的接枝在了SPVA/BC表面,吸附剂GLA-SPVA/BC成功制备。图2c为GLA-SPVA/BC吸附Cu(Ⅱ)后的材料表面,图中GLA-SPVA/BC-Cu (Ⅱ)表面的空隙变得更加的紧密。此外,图2c1为材料GLA-SPVA/BC-Cu (Ⅱ)的EDS图像,图中明显可见Cu元素的谱带,并且图2c5为Cu元素的mapping图像,Cu元素分布均匀,这表明吸附剂GLA-SPVA/BC表面具有均匀的吸附位点。
-
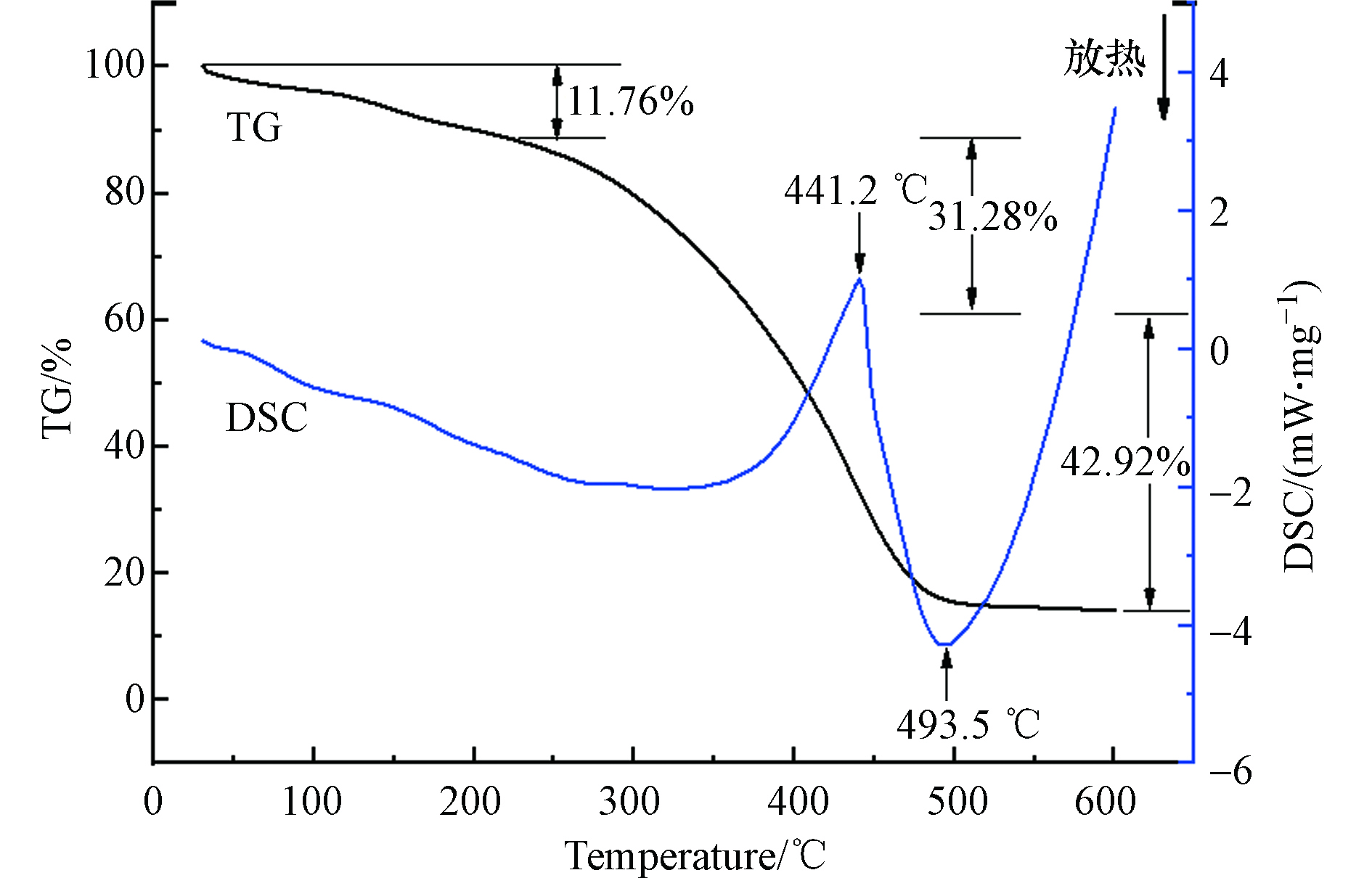
吸附剂GLA-SPVA/BC的TG-DSC分析曲线如图3所示,吸附剂GLA-SPVA/BC的失重分为3个阶段。第一阶段出现在30 —225 ℃的范围内,重量大约减少了11.76%,这可能是由于材料中的水分蒸发引起的;第二个损失阶段是225 —385.5 ℃,重量大幅度降低约31.28%,原因可能是接枝聚合物中配体的分解[12];最后一个失重阶段为385.5 —500 ℃,质量损失为42.92%,可能是由于放热链分解反应,500 ℃后,不再明显分解。吸附剂GLA-SPVA/BC的DSC曲线在441.2 ℃出现了一个强的吸收峰,可能的原因是SPVA/BC复合物的热分解放热导致的[13];强的放热峰出现在493.5 ℃,这是由于PVA链上烯基的降解导致的。由分析可知,在100 ℃以下进行吸附实验时,环境温度对材料稳定性没有影响。
-
溶液的初始pH值对吸附有着重要影响。如图4(a)所示,GLA-SPVA/BC的吸附容量明显高于SPVA/BC。随着pH的增加,SPVA/BC和GLA-SPVA/BC的吸附量先增加,然后略有下降,并在pH=5时达到最大。当pH值小于5时,随着pH的降低,由于吸附液中的H+逐渐增加,H+会与Cu(Ⅱ)竞争吸附位点,从而导致SPVA/BC和GLA-SPVA/BC的吸附能力下降,从图4(b)也可以得到证实,吸附剂GLA-SPVA/BC的zeta电位随着pH值的增加而降低。吸附剂GLA-SPVA/BC (pHzpc)的零点电荷为4.28(在pHzpc时,吸附剂表面具有零电势电荷),说明在pH值低于4.28时,吸附剂GLA-SPVA/BC表面带正电荷,吸附剂GLA-SPVA/BC与Cu(Ⅱ)的斥力降低了吸附能力。但是对于GLA-SPVA/BC,因为含硫官能团的存在,含硫基团与Cu(Ⅱ)的络合能力强,吸附量虽有下降,但是仍然体现出较大的吸附值。pH值大于4.28时,吸附剂GLA-SPVA/BC表面带负电荷,增加了吸附剂与Cu(Ⅱ)之间的静电引力,使得吸附量增大。但是pH值大于5时,随着pH值的进一步增大,会形成Cu(OH)2沉淀,改变吸附效果造成吸附量增大的假象。综上,pH值为5是吸附溶液的最佳吸附pH值。
-
分别对吸附剂SPVA/BC和吸附剂GLA-SPVA/BC的动力学数据进行了非线性的准一级动力学、准二阶动力学和伊洛维奇动力学模型拟合,此外还进行了分子内扩散模型的拟合,拟合结果分别如图5和图6所示,以此来探究吸附反应真实的吸附过程。
(1)准一级动力学方程如下:
式中,qt为t时刻吸附剂的吸附容量,mg·g−1;qe为反应达平衡时吸附剂的吸附容量,mg·g−1;k1为准一级动力学模型的速率常数,min−1。
(2)准二级动力学方程如下:
式中,k2为准二级动力学模型速率常数,g·mg−1·min−1。
(3)Elovich动力学方程如下:
式中,a为化学吸附速率常数,mg·g−1·min−1;b为复盖面积常数,g·mg−1。
(4)颗粒内扩散模型:
式中,ki为颗粒内扩散模型速率常数,mg·g−1·min−1/2;c为受吸附剂边界层影响的截距。
综合了这3个动态模型的相关参数,结果如表1所示。
由图5可知,吸附剂SPVA/BC和GLA-SPVA/BC的吸附过程可以由Elovich动力学模型更好地拟合(R2,见表1),推测吸附剂表面发生了化学吸附作用。颗粒内扩散模型拟合线段可以明显地分为两个阶段,在第一个阶段中,Cu(Ⅱ)可以比较容易的克服阻力从而通过吸附剂外表面的边界层,此阶段由于吸附剂表面拥有丰富的吸附位点,因此扩散速率较高。在第二阶段,随着吸附剂外表面逐渐饱和,Cu(Ⅱ)将逐渐扩散至吸附剂的内部,并且扩散阻力也将逐步增大。
-
对吸附剂SPVA/BC和GLA-SPVA/BC的热力学数据进行了Langmuir和Freundlich等温吸附模型的非线性拟合,拟合结果分别如图7所示,并综合了相关参数,结果见表2。
1) Langmuir吸附等温模型:
式中,qe为平衡吸附容量,mg·g−1;qm为饱和吸附容量,mg·g−1;Ce为吸附达到平衡时的吸附质的浓度,mg·L−1;KL为Langmuir吸附常数,L·mg−1。
2) Freundlich等温吸附模型:
式中,n与KF(mg·g−1)为Freundlich模型的吸附常数。
由图7可知,吸附剂SPVA/BC和GLA-SPVA/BC的Langmuir相关系数(R2,见表2)均高于Freundlich相关系数,因此吸附过程可以更好地由Langmuir等温吸附模型来拟合,表明吸附剂表面活性位点是均匀分布的,这一点与SEM中mapping图形的观察到的现象一致,且该吸附过程为单分子层吸附。由Langmuir等温吸附模型预测可知,GLA-SPVA/BC吸附剂在30 ℃的最大吸附容量为91.86 mg·g−1.
-
分别对SPVA/BC与GLA-SPVA/BC的吸附数据进行热力学参数的计算,热力学参数ΔH0,ΔS0与ΔG0可由下列公式计算得出,得出的数据如表3。
式中,KC为平衡常数;R为通用气体常数;ΔH0为焓变,J·mol−1;ΔS0为熵变J·mol−1·K−1;ΔG0为吉布斯自由能变J·mol−1。
由表3可知,ΔS0在3个温度下的值均大于0,这表明以上两种吸附剂对Cu(Ⅱ)的吸附过程是一个熵增过程,在固/液界面的混乱度不断增加。对GLA-SPVA/BC来说,其对Cu(Ⅱ)的吸附过程是自发的(ΔG<0),反应体系不需从外界吸收热量。此外,两者的焓变均为正值,表明其吸附过程为吸热的,因此,较高的温度可以提高材料的吸附性能。与此同时,GLA-SPVA/BC包含官能团,如C=S和氨基,允许它进行化学吸附,由焓变化数据证实。由于醛基氧化为羧基离子,SPVA/BC进行了金属离子的化学吸附,这也证实了pH的变化对其吸附能力的影响。
-
吸附剂GLA-SPVA/BC对Cu(Ⅱ)的吸附机理可通过氨基和含硫官能团的配位作用(主要作用)以及表面静电吸附作用进行解释。吸附剂GLA-SPVA/BC中的脒基硫脲含硫官能团=S和含N官能团,对Cu(Ⅱ)表现出配位作用(图8)。
此外,由zeta电位(图4(b))可以看出,当pH=5.0时,GLA-SPVA/BC表面带负电荷,表明吸附剂与Cu(Ⅱ)之间存在静电引力。为了证实GLA-SPVA/BC对金属离子的配位机理,对其吸附铜离子前后的材料进行了XPS光谱分析,如图8所示。从图8(b)中可以看出,吸附Cu(Ⅱ)之后,除了材料(图8(a)所示)原有的4个峰外,还有一个新的峰在932.94 eV出现,证实了Cu(Ⅱ)成功的被吸附。此外,Cu(Ⅱ)吸附前后N1s的XPS光谱如图(c)和(d)所示,吸附前,可以将3个峰(398.48、399.61、400.06 eV)分配给C=N、C—N—C和—NH2[14];吸附后,这3个峰稍微位移到398.81、400.01、400.83 eV,这是由于GLA-SPVA/BC中N和金属离子之间的配位相互作用造成的[15]。GLA-SPVA/BC吸附前的S2p谱如图(e)和(f)所示,可以看出吸附前原本有3个峰[16],164.16/165.54 eV(C=S/C—S)和161.81 eV(S—H);吸附后,C=S/C—S的峰值有所降低,可能是电子从C=S转移到Cu(II)引起的。XPS的分析结果进一步证实了GLA-SPVA/BC吸附重金属离子主要依赖于材料中的硫脲基官能团,说明吸附是以N和S的配位作用为主、静电吸附为辅的作用机理,吸附示意图如图9(b)所示。
为研究吸附剂GLA-SPVA/BC的吸附选择性,实验选取了3种与Cu(Ⅱ)相似的金属离子为竞争离子,其选择性吸附如图9(a)所示,可以看出GLA-SPVA/BC吸附剂对Cu(Ⅱ)表现出高选择性。根据软硬酸碱理论,GLA-SPVA/BC中脒基硫脲的含硫官能团=S属于软碱,而这四种金属均属于交界酸,所生成的配合物稳定性相差不大,因此无法解释选择性的差异。造成这一现象可能与吸附剂上的氨基有关,原因有三:①金属离子电子结构[17],Ni(Ⅱ)(3s23p63d8)、Cu(Ⅱ)(3s23p63d9)、Zn(Ⅱ)(3s23p63d10)、Pb(Ⅱ)(5s25p65d106s2),从Ni(Ⅱ)到Zn(Ⅱ)和Pb(Ⅱ)的d轨道由未充满到全充满,电子云密度增大,配体向中心离子转移电子程度依次减小,中心离子接受配体的配位电子的能力依次减弱,导致-NH2对Cu(Ⅱ)、Pb(Ⅱ)、Ni(Ⅱ)和Zn(Ⅱ)的作用力依次减弱。②根据Irving-Williams序列和Jahn-Teller效应,镍、铜和锌其二价金属离子在与含N配位原子的配体生成的配合物时,稳定次序是Ni(Ⅱ)<Cu(Ⅱ)>Zn(Ⅱ),也体现出对Cu(Ⅱ)的较强作用力。③与Pb(Ⅱ)半径(1.19)[18]相比,Cu(Ⅱ)半径(0.73)小,吸引电子的能力大,更容易吸引氨基的孤对电子对形成配位键。综上原因,吸附剂GLA-SPVA/BC在4元溶液中对Cu(Ⅱ)体现出明显的选择性。
-
本研究用脒基硫脲改性交联聚乙烯醇/细菌纤维素(SPVA/BC)制备了一种新型吸附剂(GLA-SPVA/BC),该吸附剂对Cu(Ⅱ)具有较高的吸附量和优异的选择性。用SEM/ EDS-Mapping、FT-IR光谱等对材料进行了表征,证实了脒基硫脲在SPVA/BC表面的成功接枝;XPS光谱以及选择性实验的结果表明GLA-SPVA/BC对铜离子是以S和N配位作用为主,并在四元体系中表现出较高的选择性。Elovich动力学模型较好地拟合了GLA-SPVA/BC吸附剂的吸附动力学数据;Langmuir等温吸附模型预测GLA-SPVA/BC吸附剂在30 ℃时的最大吸附容量为91.86 mg·g−1。该吸附剂的合成为选择性吸附Cu(Ⅱ)提供了新的思路方法。
脒基硫脲接枝聚乙烯醇/细菌纤维素吸附剂的制备及其对Cu(Ⅱ)的选择性吸附
Preparation of amidyl thiourea grafted polyvinyl alcohol/bacterial cellulose adsorbent and its selective adsorption of Cu(Ⅱ)
-
摘要: 本研究将脒基硫脲(GLA)接枝到交联聚乙烯醇(SPVA)/细菌纤维素(BC)制备了一种新吸附剂(GLA-SPVA/BC),并研究其对铜离子的选择性吸附。FT-IR中C=N和—(C=S)—N特征吸收峰的出现以及mapping图像中N和S元素的均匀分布表明脒基硫脲成功接枝在SPVA/BC表面;TG-DSC的结果表明吸附剂GLA-SPVA/BC在吸附过程中具有很好的热稳定性。该吸附剂GLA-SPVA/BC对Cu(Ⅱ)离子的吸附符合Langmuir等温线模型,30 ℃时最大吸附容量为91.86 mg·g−1;Elovich动力学模型拟合显示在吸附剂表面发生了化学吸附作用;XPS谱图中N、S特征峰值的变化进一步证实是主要通过配位作用完成吸附。GLA-SPVA/BC吸附剂在铜锌铅镍4元离子体系中,对Cu(Ⅱ)表现出高的选择性。Abstract: In this study, we studied that the amidyl thiourea (GLA) was grafted to the matrix of cross-linked polyvinyl alcohol (SPVA) and bacterial cellulose (BC) to prepare a new adsorbent (GLA-SPVA/BC), selectively adsorbing copper ions. The C=N and —(C=S)—N characteristic absorption peaks in FT-IR spectrum of GLA-SPVA/BC and the uniform distribution of N and S elements in the element mapping image confirmed that GLA was successfully grafted onto the surface of SPVA/BC. The results of TG-DSC showed that GLA-SPVA /BC had good thermal stability during the adsorption process. The adsorption of Cu(Ⅱ) ions by the adsorbent GLA-SPVA/BC complies with the Langmuir isothermal model, and there is a maximum adsorption capacity of (91.86 mg·g−1) at 30 ℃. The fitting dynamical model verified a chemo-adsorption on the surface of the GLA-SPVA/BC. The adsorption by coordination way was confirmed by the change of N1s and S1s characteristic peak in the XPS spectrum. GLA-SPVA/BC shown high selectivity to Cu(Ⅱ) in Cu-Zn-Pb-Ni 4 ion system.
-

-
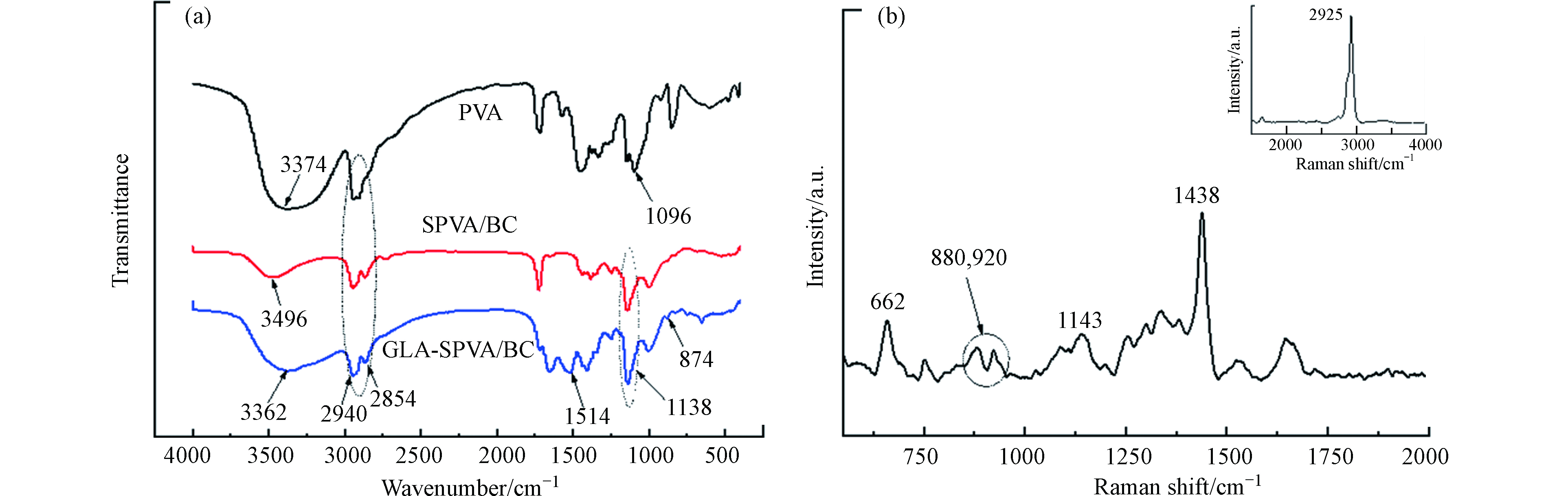
图 1 (a)PVA、SPVA/BC和GLA-SPVA/BC的红外吸收谱图和(b) GLA-SPVA/BC的拉曼光谱图
Figure 1. (a) Infrared absorption spectra of PVA SPVA/BC and GLa-SPVA /BC and (b) Raman spectra of GLa-SPVA /BC

图 2 (a)SPVA/BC、(b) GLA-SPVA/BC和 (c)GLA-SPVA/BC-Cu (Ⅱ)的SEM图像;(b1—b4) GLA-SPVA/BC的EDS-mapping图像;(c1—c5)GLA-SPVA/BC-Cu (Ⅱ)的EDS-mapping图像
Figure 2. (a) SEM images of SPVA / BC, (b) GLA-SPVA / BC, and (c) GLA-SPVA / BC-Cu (Ⅱ); (b1—b4) EDS-mapping images of GLA-SPVA / BC; and (c1—c5) EDS-mapping images of GLA-SPVA / BC-Cu (Ⅱ)
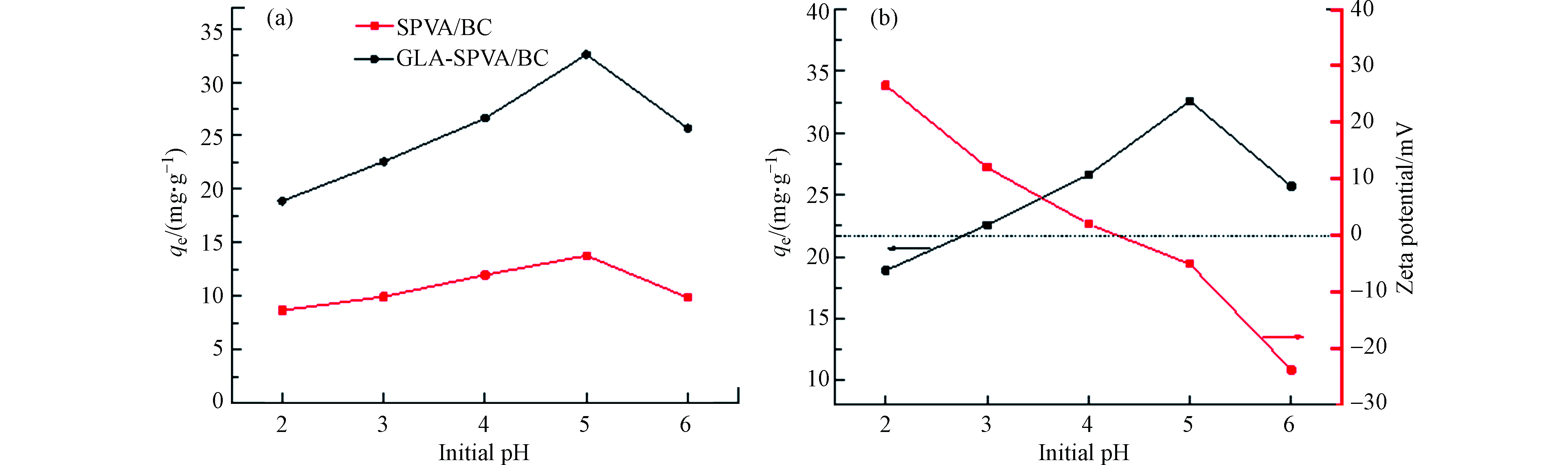
图 4 (a)溶液的初始pH值对Cu(Ⅱ)吸附容量的影响;(b)不同pH对吸附剂的zeta电位和吸附能力的影响
Figure 4. (a) The effect of initial pH value on the adsorption capacity of Cu(Ⅱ);(b) The effect of different pH on zeta potential and adsorption capacity of adsorbent
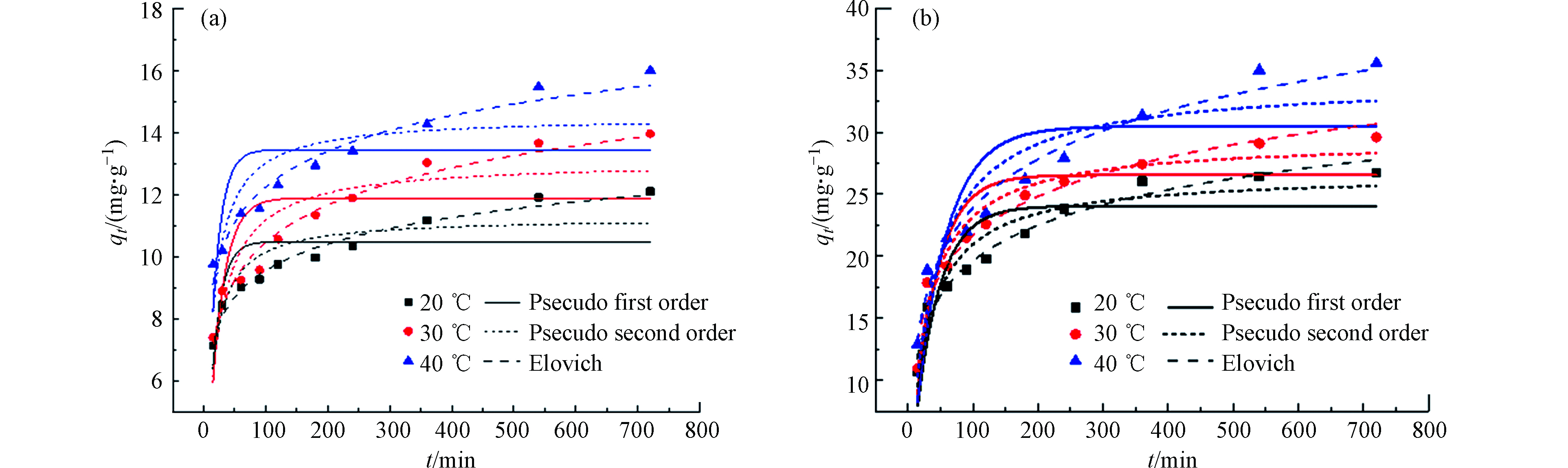
图 5 (a) SPVA/BC和(b) GLA-SPVA/BC的非线性动力学模型
Figure 5. (a) SPVA/BC and (b) GLA-SPVA /BC nonlinear dynamics models
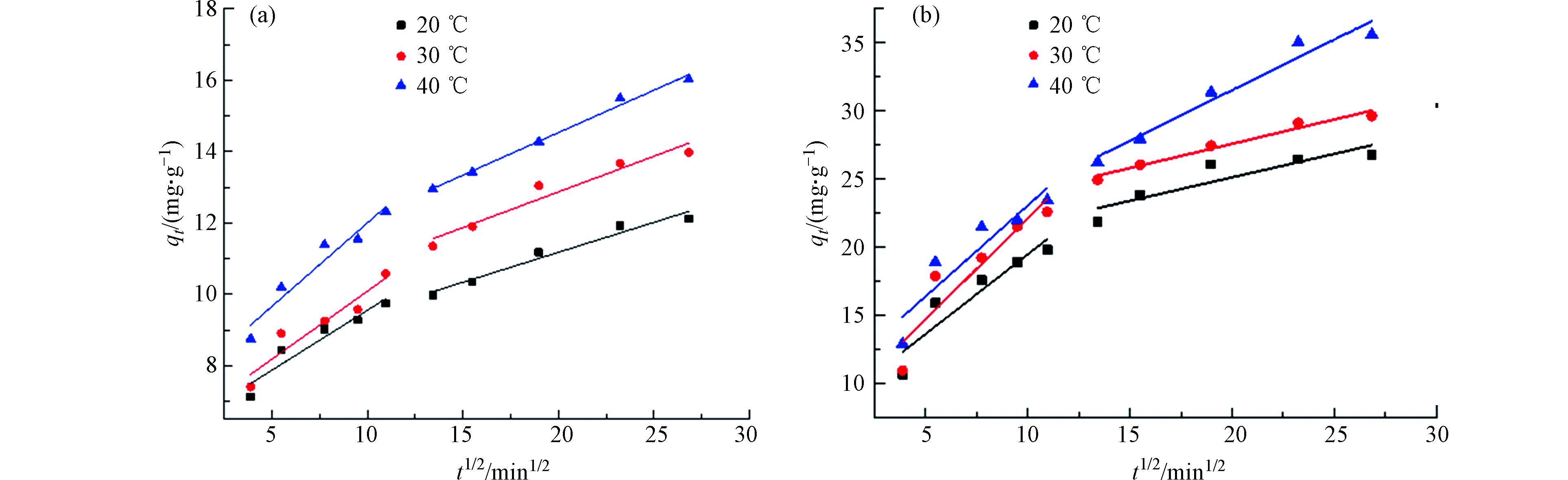
图 6 (a) SPVA/BC和(b) GLA-SPVA/BC的颗粒内扩散模型
Figure 6. (a) SPVA/BC and (b) GLA-SPVA /BC intra particle diffusion models

图 7 (a) SPVA/BC 吸附剂与(b) GLA-SPVA/BC吸附剂在不同温度下的热力学拟合图
Figure 7. (a) Thermodynamic fitting diagram of SPVA/BC adsorbent and (b) GLa-SPVA /BC adsorbent at different temperatures

图 8 GLA-SPVA/BC吸附Cu(Ⅱ)离子前后的高分辨率XPS光谱
Figure 8. High resolution XPS spectra of Gla-SPVA /BC before and after adsorption of Cu(Ⅱ) ions: Total spectra (a) and (b) N1s(c) and (d) S2p(e) and (f)
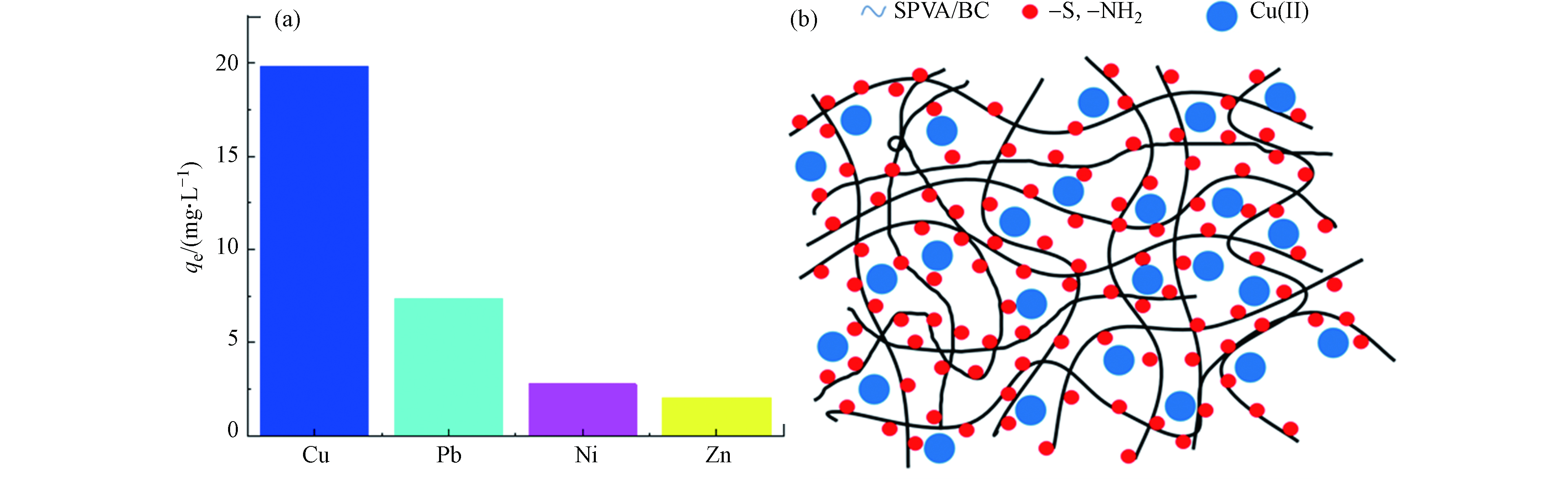
图 9 (a)吸附剂GLA-SPVA/BC在四元共混体系中的选择性吸附;(b )吸附示意图
Figure 9. Schematic diagram of selective adsorption and adsorption of GLA-SPVA /BC in the quaternary blend system
表 1 准一级动力学模型与准二级动力学模型动力学参数
Table 1. Kinetic parameters of quasi-first-order and quasi-second-order kinetic models
吸附剂
AdsorbentT/℃ qe,exp/
(mg·g−1)Pseudo-first-order model Pseudo-second-order model Elovich kinetic model qe,cal/
(mg·g−1)k1/
min−1R2 SD/% qe,cal/
(mg·g−1)k2/
(g·mg−1·min−1)R2 SD/% qe,cal/
(mg·g−1)a/
(mg·g−1·min−1)b/
(g·mg−1)R2 SD/% SPVA/
BC20 13.38 12.26 0.0113 0.8704 21.22 14.01 0.0011 0.9476 14.12 13.31 0.619 0.3868 0.9820 7.53 30 17.97 15.10 0.0270 0.6882 15.11 16.79 0.0022 0.8867 9.09 17.50 2.823 0.3799 0.9890 2.31 40 24.78 20.71 0.0291 0.6069 16.15 22.97 0.0017 0.8414 10.43 24.04 4.664 0.2857 0.9810 1.66 GLA-
SPVA/
BC20 18.2 16.67 0.0069 0.9177 24.01 20.22 0.0004 0.9492 19.17 16.80 0.452 0.2653 0.9366 19.39 30 30.18 25.49 0.0173 0.6639 21.45 28.31 0.0009 0.8510 14.08 28.64 2.735 0.2104 0.9684 6.06 40 36.05 32.47 0.0106 0.7590 25.54 36.87 0.0004 0.8714 18.89 34.62 1.785 0.1525 0.9438 11.67  下载: 导出CSV
下载: 导出CSV
表 2 吸附剂吸附Cu(Ⅱ)的等温线参数
Table 2. Isotherm parameters of adsorption of Cu(Ⅱ) by adsorbent
吸附剂
AdsorbentT/°C Langmuir models Freundlich models qm/(mg·g−1) KL/(L·mg−1) R2 KF/(mg·g−1) 1/n R2 SPVA/BC 20 50.81 0.0029 0.9414 0.376 0.733 0.9167 30 69.11 0.0039 0.9839 0.857 0.700 0.965 40 78.52 0.0070 0.9992 2.453 2.453 0.9812 GLA-SPVA/BC 20 66.41 0.0031 0.9583 0.535 0.724 0.9293 30 82.36 0.0051 0.9958 1.545 0.620 0.9755 40 86.57 0.0146 0.9881 6.592 0.433 0.9905
下载: 导出CSV
表 3 Cu (Ⅱ)吸附于吸附剂上的热力学参数
Table 3. Thermodynamic parameters of Cu(Ⅱ) adsorbed on an adsorbent
吸附剂
Adsorbent温度T/K 吉布斯自由能ΔG0/(kJ·mol−1) 焓变ΔH0/(kJ·mol−1) 熵变ΔS0/(J·mol−1·K−1) SPVA/BC 293.15 -12.35 19.97 110.24 303.15 −13.45 313.15 −14.55 GLA-SPVA/BC 293.15 −15.12 7.35 76.64 303.15 −15.88 313.15 −16.65
下载: 导出CSV
-
[1] REYES-SERRANO A, LÓPEZ-ALEJO J E, HERNÁNDEZ-CORTÁZAR M A, et al. Removing contaminants from tannery wastewater by chemical precipitation using CaO and Ca(OH)2 [J]. Chinese Journal of Chemical Engineering, 2020, 28(4): 1107-1111. doi: 10.1016/j.cjche.2019.12.023 [2] MOGHIMI F, JAFARI A H, YOOZBASHIZADEH H, et al. Adsorption behavior of Sb(III) in single and binary Sb(III)—Fe(II) systems on cationic ion exchange resin: Adsorption equilibrium, kinetic and thermodynamic aspects [J]. Transactions of Nonferrous Metals Society of China, 2020, 30(1): 236-248. doi: 10.1016/S1003-6326(19)65195-2 [3] XIAO D, DING W, ZHANG J B, et al. Fabrication of a versatile lignin-based nano-trap for heavy metal ion capture and bacterial inhibition [J]. Chemical Engineering Journal, 2019, 358: 310-320. doi: 10.1016/j.cej.2018.10.037 [4] AYDIN Y A, AKSOY N D. Adsorption of chromium on chitosan: Optimization, kinetics and thermodynamics [J]. Chemical Engineering Journal, 2009, 151(1/2/3): 188-194. [5] WANG X H, YANG L, ZHANG J P, et al. Preparation and characterization of chitosan-poly(vinyl alcohol)/bentonite nanocomposites for adsorption of Hg(II) ions [J]. Chemical Engineering Journal, 2014, 251: 404-412. doi: 10.1016/j.cej.2014.04.089 [6] TRIKKALIOTIS D G, CHRISTOFORIDIS A K, MITROPOULOS A C, et al. Adsorption of copper ions onto chitosan/poly(vinyl alcohol) beads functionalized with poly(ethylene glycol) [J]. Carbohydrate Polymers, 2020, 234: 115890. doi: 10.1016/j.carbpol.2020.115890 [7] WU Z Y, LIANG H W, CHEN L F, et al. Bacterial cellulose: A robust platform for design of three dimensional carbon-based functional nanomaterials [J]. Accounts of Chemical Research, 2016, 49(1): 96-105. doi: 10.1021/acs.accounts.5b00380 [8] XU X R, CHEN X, YANG L Y, et al. Film-like bacterial cellulose based molecularly imprinted materials for highly efficient recognition and adsorption of cresol isomers [J]. Chemical Engineering Journal, 2020, 382: 123007. doi: 10.1016/j.cej.2019.123007 [9] 刘海龙, 何璐红, 赵扬. 硫脲基重金属离子吸附材料的研究进展[C]//第三届河南省化学、化工与生物、食品学术研讨会论文集. 平顶山市, 2015: 26-29. LIU H L, HE L H, ZHAO Y. Research progress of thiourea - based heavy metal ion adsorption materials[C]//Proceedings of the 3rd Henan Provincial Symposium on Chemistry, Chemical engineering and Biological Food. Pingdingshan City, 2015: 26-29(in Chinese).
[10] MONIER M, ABDEL-LATIF D A. Modification and characterization of PET fibers for fast removal of Hg(II), Cu(II) and Co(II) metal ions from aqueous solutions [J]. Journal of Hazardous Materials, 2013, 250/251: 122-130. doi: 10.1016/j.jhazmat.2013.01.056 [11] SONG L, SHU L, WANG Y Q, et al. Metal nanoparticle-embedded bacterial cellulose aerogels via swelling-induced adsorption for nitrophenol reduction [J]. International Journal of Biological Macromolecules, 2020, 143: 922-927. doi: 10.1016/j.ijbiomac.2019.09.152 [12] WANG S, GAO Q Y, WANG J C. Thermodynamic analysis of decomposition of thiourea and thiourea oxides [J]. The Journal of Physical Chemistry B, 2005, 109(36): 17281-17289. doi: 10.1021/jp051620v [13] 张洪玉, 杨亮, 陆大年. 细菌纤维素基聚乙烯醇(BC/PVA)复合膜的制备及性能研究 [J]. 印染助剂, 2012, 29(8): 18-21. doi: 10.3969/j.issn.1004-0439.2012.08.005 ZHANG H Y, YANG L, LU D N. Preparation and properties of polyvinyl alcohol (PVA) composites membranes based on bacterial cellulose (BC) [J]. Textile Auxiliaries, 2012, 29(8): 18-21(in Chinese). doi: 10.3969/j.issn.1004-0439.2012.08.005
[14] AWAD F S, ABOUZEID K M, EL-MAATY W M A, et al. Efficient removal of heavy metals from polluted water with high selectivity for mercury(II) by 2-imino-4-thiobiuret-partially reduced graphene oxide (IT-PRGO) [J]. ACS Applied Materials & Interfaces, 2017, 9(39): 34230-34242. [15] CHEN Z C, TANG B T, NIU Y Z, et al. Synthesis of silica supported thiosemicarbazide for Cu(II) and Zn(II) adsorption from ethanol: A comparison with aqueous solution [J]. Fuel, 2021, 286: 119287. doi: 10.1016/j.fuel.2020.119287 [16] LIU P, WANG X L, TIAN L, et al. Adsorption of silver ion from the aqueous solution using a polyvinylidene fluoride functional membrane bearing thiourea groups [J]. Journal of Water Process Engineering, 2020, 34: 101184. doi: 10.1016/j.jwpe.2020.101184 [17] 李怀娜, 尤进茂, 李峰, 等. 乙醛酸缩氨基硫脲和锌(Ⅱ)、铅(Ⅱ)、铜(Ⅱ)、锰(Ⅱ)、镍(Ⅱ)、钴(Ⅱ)配合物的薄层色谱与紫外光谱的研究 [J]. 色谱, 1995, 13(3): 203-204. LI H N, YOU J M, LI F, et al. A study on the thin-layer chromatography and ultraviolet spectrum of cobalt(Ⅱ), copper(Ⅱ), zinc(Ⅱ), lead(Ⅱ), manganese(Ⅱ)and nickel(Ⅱ)as their glyoxylic acid thiosemicarbazone complexes [J]. Chinese Journal of Chromatography, 1995, 13(3): 203-204(in Chinese).
[18] SHANNON R D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides [J]. Acta Crystallographica Section A, 1976, 32(5): 751-767. doi: 10.1107/S0567739476001551 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 2326
- HTML全文浏览数: 2326
- PDF下载数: 105
- 施引文献: 0
