
















-
全氟和多氟烷基化合物(per- and polyfluoroalkyl substances,PFAS)是一类重要的化学品,碳链上的氢原子被部分或者全部取代为氟原子,其物化性质与结构如表1所示. PFAS按照其碳链长度可分为短链和长链PFAS,并按照其分子量的差异以气体、液体或固体高分子聚合物等方式存在. 全氟羧酸类化合物(PFCAs)、全氟磺酸类化合物(PFSAs)和全氟磺酰胺类化合物(FASAs)具有相似的全氟碳链结构,但含有不同的末端官能团,分别为—COOH、—SO3H和 —SO2—N—CH2CH3. 此外,多氟调聚醇类化合物(FTOHs)的结构式为F(CF2)nCH2CH2OH,可以作为聚合物和表面活性剂中间产物的合成原料. PFAS具有良好的疏水性、疏油性和抗污性,由于其C—F具有较强的键能(约536 kJ·mol−1)[1],比碳氢有机化合物具有更稳定的化学性质,被广泛运用于化工、服装家具、食品包装和日常用品等行业中,最新研究发现PFAS还可运用于弹药、人造草皮和土壤修复等方面中[2].
PFAS的主要来源包括垃圾填埋场、水成膜泡沫 (AFFF)、金属电镀和汽车/金属冲压,这4个来源占污染总量的75%. 在不同的PFAS来源附近,环境介质中PFAS的赋存状态也会有一定的区别. 在垃圾填埋场收集到的渗透液和污水处理厂流出废流中,最主要的PFAS为PFOA、PFOS和短链化合物(PFHxA、PFBA和PFBS),除此之外生物转化产物氟调聚物羧酸(FTCAs)和全氟烷基酸(PFAAs)也常被检测到. 垃圾填埋厂渗透液中PFAS总浓度范围为0.2—45.6 μg·L−1[3],而污水处理厂废水和污泥中PFAS含量分别为21—560 ng·L−1和3.2—150 ng·g−1[4]. AFFFs可用来扑灭碳氢类燃料引起的火灾,因此被常用于机场和军事的消防演习中,在美国某军事基地的地下水中,检测到PFAS的浓度范围为0.5—
1478 μg·L−1[5]. 含氟化学品制造设施的排放废水也是PFAS的直接来源之一,由于PFAS使用的禁止,氟类替代品逐渐取代了PFAS在工业中的使用地位并逐渐成为一类重要的污染物. 在含氟化学品制造厂附近的地表水中,PFCAs和PFSAs的总浓度为0.35 μg·L−1,而氟化替代品全氟2-甲基-3-氧杂己酸 (PFPrOPrA)的浓度达到了4.5 μg·L−1[6]. 在纺织和纸张涂料行业,由于替代品氟乙烯基聚合物的使用,PFOS的含量逐渐减少. 然而,PFOS及其前体物质全氟辛烷磺酰胺乙醇基磷酸酯(SAmPAPs)仍然是污染水中主要的PFAS,在造纸厂附近湖泊的沉积物中检测到的PFAS浓度为2450 μg·g−1[7]. 在金属电镀行业,含PFOS的抑雾剂及其替代品F-53B(6:2氯代氧杂氟烷磺酸钾,商业名称)也成为了污染水中较为主要的化学物质,在镀铬电镀厂的排出废水中检测到PFOS和F-53B的浓度分别为44.1 μg·L−1和102.9 μg·L−1[8].PFAS的物理化学性质稳定,具有难降解性、蓄积性和生物毒性,并可以通过多种环境介质进行长距离的迁移,在环境[9]、动物[10 − 11]和人体[12 − 13]中被频繁检测到. 全氟类化合物进入人体后,会与血清蛋白结合[12]沉积在脏器中,引起癌细胞的生成[13]并造成血脂异常[14],同时还能引起甲状腺异常[15]和免疫反应[16],并影响生殖健康. 此外,母乳中也能检测到PFAS的存在[17],其中全氟辛烷磺酸(PFOS)和全氟辛烷羧酸(PFOA)的含量分别达到了321 pg·mL−1和411 pg·mL−1. 此外PFAS在饮用水、工业废水等多种环境介质中检出 [6, 18],在水体中(以珠江和深圳河为例),PFAS的含量超过了10 ng·L−1;在远离城市的自然保护区水体中,PFAS平均最大值为1.3 ng·L−1[19]. PFAS作为持久性有机污染物(persistent organic pollutants,POPs)中的一种,可以通过水体或大气等环境介质进行迁移,发生PFAS的全球性扩散. 2010年在青藏高原的气体中检测到了PFAS的痕迹[20],可能是来源于东南亚的人类活动和印度洋上海洋气溶胶积雨沉降[21]. 此外,在室内和室外的粉尘中也检出了PFAS的存在,自2013到2017年,中国室外粉尘中PFAS浓度从63 ng·g−1升高至164 ng·g−1[22].
PFOA和PFOS这两类特征PFAS化合物在环境中的检出较多,并在2009年被《斯德哥尔摩公约》附件B列为新POPs. 欧盟在2020年7月发布的持久性污染物法规表明要限制PFOA及其化合物的使用,而中国作为氟类化合物的最大生产国和使用国,在2022年3月《生活饮用水卫生标准》中规定PFOA和PFOS的限值为80 ng·L−1和40 ng·L−1. 随着PFOA和PFOS被限制,短链含氟化合物和氟替代品逐步替代中长链含氟化合物[23]在工业生产中的重要地位,但这些化合物同样具有强的C—F键能,表现出与传统的中长链含氟化合物相似的物理化学性质. 近几年来,国际上对多氟类化合物的危害也越来越重视,美国加州制定法规限制了多氟类化合物的使用[24]. 可见,PFAS对生态系统和人类健康都造成了一定的潜在危险,因此寻找绿色环保的替代化合物,和探索节能高效的PFAS去除新技术已成为当今世界各国亟需解决的环境科学问题. 高级氧化法、高级还原法和微生物降解法是目前在PFAS的去除领域中最主要的方法. 本文综述了过硫酸盐活化、电化学氧化、声化学降解、光催化法、高级还原法、微生物降解法的去除途径,去除机理,去除特点、去除效果,并分析其优点及不足之处,对未来全氟和多氟烷基化合物的去除技术的发展和应用前景提出展望.
-
高级氧化法(advanced oxidation processes,AOPs)作为一种较为新兴的氧化技术,具有氧化能力强、无选择性、反应速度快和处理效率高等特点被广泛应用. 高级氧化法通过产生具有高活性的氧化物种(SO4•−、•OH、1O2等)来实现对有机类污染物的高效去除. 去除PFAS的AOPs技术中,常用的有热活化过硫酸盐法、电化学氧化法、声化学氧化法和光催化法.
-
热活化过硫酸盐法是目前处理PFAS使用较为广泛的高级氧化法. 过硫酸盐加热到一定的温度时,其会被活化产生硫酸根自由基(SO4•−)(式1),并与目标污染物发生单电子转移,实现污染物的降解[25]. 以PFOA为例,SO4•−先与PFOA发生电子转移形成PFOA自由基(式2),PFOA自由基脱羧后生成全氟烷基自由基(C7F15•)(式3). C7F15•有多种反应路径,可以和水进行羟基化反应(式4),也可以与氧气结合,通常认为C7F15•与氧气结合生成全氟烷基过氧自由基C7F15OO•,并经过双分子衰变最终形成C7F15O•(式5、式6). C7F15O•进一步与溶液中的HSO4−反应生成C7F15OH(式7),最终通过热转化和水解生成C6F13COOH(式8、式9),从而减少一个CF2结构单元. [26] SO4•−通过这种逐步分解的反应机制来实现对PFOA的矿化(图1).
通过水热、微波、光照、Fe2+催化剂等方法都可以活化过硫酸盐产生SO4•−,进而用于 PFAS的去除. Yang等[27]发现热活化对PFOA具有很好的去除率. 在降解的过程中,溶液pH会随着时间的增大而减小(式10). 但实验表明,反应过程中的pH并不会对PFOA的脱氟率产生较大影响,脱氟率更多取决于反应的初始pH. 酸性体系中H+能催化过硫酸盐产生SO4•− (式11、式12);而在碱性体系中,SO4•−会进一步与体系中的OH−发生反应,生成•OH和SO42− (式13)[28],从而影响PFOA去除率.
过硫酸盐的起始浓度也会影响PFOA的去除率. 随着过硫酸盐浓度增加,PFOA脱氟率先增大后逐渐降低,当溶液中过硫酸盐的浓度达到饱和时,SO4•−会与过硫酸盐、SO4•−[29 − 30]、水[31]等发生系列副反应(式14、式16),导致其与PFAS反应的选择性降低[28],从而降低了PFOA的去除率.
Liu等[32]研究了热活化温度对PFOA去除率的影响. 在过硫酸盐的体系中,温度的升高促进了SO4•−的产生,经过30 h反应后,PFOA去除率从0%升高至93.5%,脱氟率达到了43.6%. 此外,该团队进一步研究了同时使用热活化和Fe2+催化过硫酸盐时对PFOA降解性能的影响,结果表明加入Fe2+反而降低了PFOA去除率,这主要是因为Fe2+可以活化过硫酸盐 (式17)[33],但在高温下其反而成为了硫酸根自由基的清除剂(式18)[34],导致PFOA整体的去除率降低.
热活化法还可以结合其他高级氧化技术来提升对PFAS的降解效率(表2). Lee等[28]使用微波-热活化法降解PFOA,与传统的热活化法相比,微波-热活化法的降解速率提高了3—5倍,且能耗降低了50%,具有更大的发展潜力. 在实际应用中,还可对体系进一步改进,例如在微波的体系中再加入零价铁(ZVI)[35]. ZVI能在高温水热条件下对PFCAs进行部分降解[36]并产生Fe2+,进而催化活化过硫酸盐产生SO4•−,促进PFOA的分解[31]. 因此,热活化法不仅能有效的活化过硫酸盐产生SO4•−,还能加快PFOA降解反应的热力学过程.
热活化过硫酸盐处理PFAS是目前使用较为广泛的一种方法,其加热过程中会产生SO4•−,并通过逐步分解PFAS的反应机制实现矿化. 尽管热活化法操作简单、去除效率高. 但不可否认的是,热活化过硫酸盐产生SO4•−的性能受过硫酸盐浓度、热活化温度和溶液起始pH值的影响,从而导致PFAS去除性能不够稳定. 此外,真实水环境中Cl−等共存阴离子会与SO4•−发生副反应,影响PFAS的降解效率. 除此之外,过硫酸盐在降解PFAS过程中是反应物而不是催化剂,在反应中会被持续消耗,因此在实际水处理过程中需要不断添加过硫酸盐并维持稳定的活化温度,从而实现PFAS的彻底矿化. 基于此,后续的研究可以更关注:(1)与微波等方法结合使用,提高降解效率;(2)优化反应体系,拓宽可适用的pH范围;(3)寻找优异的催化剂,降低过硫酸盐浓度和活化温度;(4)实际水体处理时,可先进行预处理,减少杂质对PFAS降解的影响.
-
电化学氧化(electrochemical oxidation,EO)是指污染物在电极表面发生直接电化学反应或利用电极表面产生的具有强氧化性的活性物种进行污染物的间接电化学氧化. 电化学氧化技术是一种新颖且应用广泛的降解方法,具有操作简单、反应条件温和、无二次污染、后续处理简单等优点,并且在PFAS的处理中也表现出了高效的降解能力,成为了目前水污染控制领域的一个研究热点.
EO法降解有机污染物的反应机理可分为羟基自由基(•OH)反应和电子转移反应(direct electron transfer,DET)[37]. 由于PFAS中C—F较高的键能[38],•OH无法对PFAS进行有效的降解. 研究发现PFAS的电化学氧化降解包括3个反应步骤,首先直接在阳极表面发生电子转移[37],这不仅是整个反应的起始步骤,还是决速步骤[39]. 以PFSAs为例[40],PFSAs在阳极失去一个电子后变为全氟磺酸自由基CnF2n+1SO3•,进一步脱磺变为全氟自由基CnF2n+1•,之后再与体系中的•OH或O2反应,发生水解反应并逐步分解. 图2中展示了3种PFAS电化学氧化降解途径. 在这3种降解方法中,尽管循环1中与•OH的反应被认为是能量上最有利的反应途径,但在实际过程中全氟自由基更偏向于与浓度更大的氧气进行进一步的反应. 3种途径都需要先完成第一步的官能团脱去步骤(脱磺)[41].
Zhuo等[43]研究了溶液pH对电化学降解PFOS效率的影响. 在碱性条件下,OH−由于静电吸引,会更容易吸附在阳极表面,影响目标污染物的降解效率. 除此之外,电化学过程的电流密度也会影响去除率. 电子转移效率提高并增大析氧电位,加快电化学氧化. 但当电流密度进一步增大到一定限度时(5.88 mA·cm−2),反而促进了析氧等副反应进行,并阻碍PFOS从溶液扩散到电极表面,导致反应速率减小[44]. PFAS碳链的长度也会影响降解速率,Zhuo等[43]研究了7种不同碳链长度和种类的PFAS在BDD电极上的降解趋势. 结果表明PFAS的碳链越长,疏水性越强,越容易吸附在疏水的BDD电极上,从而具有最快的电化学降解速率. Niu等[45]使用改性的PbO2薄膜电极也证明了类似的结论,随着PFOS碳链的增长,其TOC去除率也逐渐增大. 此外PFAS的末端官能团种类也会影响其降解速率,磺基的亲电性比羧基更强,导致全氟磺酸(PFSA)更难发生失电子的电子转移过程. 因此,碳链长度相同时,电化学降解全氟羧酸(PFCA)的反应速率比PFSA更快.
在电化学氧化降解PFAS方法中,阳极材料是影响PFAS去除效率的关键因素,虽然BDD电极具有较高的析氧潜力和较强的氧化能力,但制备困难且成本较高,因此发展其他的金属氧化物阳极材料成为了研究的热点,表3总结了不同阳极材料对PFAS的电化学氧化能力,PFAS去除率主要取决于电极的电导率、羟基自由基产率、析氧电位以及阳极材料的比表面积和孔径分布等因素. 在近年来的研究中,对阳极材料和结构的改性可明显增加PFAS的去除效率. Hwang等[46]通过增大电解液与电极材料的表面接触面积,在2 h内实现了91%的PFOS降解率. 此外,将不同电极材料(Ti电极上包覆纳米氧化锌颗粒)进行复合,也可以提高电极的物理化学稳定性,从而获得更加优异的电化学氧化性能[47]. 除了改性电极,电化学氧化技术还能与水热[48]、吸附、声化学等方法进行结合,提高降解效率并减少电能消耗.
电化学氧化法可通过直接电化学氧化污染物,也可产生羟基自由基等活性物种进行间接的电化学氧化,是一种高效且绿色环保的高级氧化法. 电化学氧化过程是在电极/溶液的界面上进行,因此阳极材料的电化学性能(析氧电位、电导率、疏水性、羟基自由基生成能力等)直接影响了对PFAS降解的效果. 当电流密度增加时,电化学体系中发生的析氧副反应会影响自由基的产率,进一步影响PFAS的降解效率. 除此之外,电解质溶液种类和浓度、溶液pH、污染物种类和浓度、电解时间、电流密度等因素也会对电化学氧化体系的降解速率产生影响. PFAS的官能团种类和碳链长度会影响污染物的亲电性和疏水性,进一步影响在电极表面发生的直接电子转移过程的进行. 对电化学氧化法后续的研究可以着眼于:(1)在电极上负载或掺杂具有高电催化活性的材料,并改进制备方法,提高电极的稳定性和电化学氧化能力;(2)优化电极结构和反应器结构,增大电极/溶液界面的接触面积的同时促进传质过程的进行;(3)将电化学过程与其他过程(水热、声化学等)相结合,提高自由基产量、传质速率和吸附能力等来实现协同效应,进一步提高有机物的降解率和有机物的降解程度.
-
声化学降解技术能在接近环境温度和压力的条件下,在较短的时间内在水溶液中诱发具有高能量的化学反应. 声化学降解的基本原理是利用超声技术产生空化现象,在超声辐照下液体中产生空化气泡,空化气泡生长后发生内爆塌缩[52],造成局部高温高压[53]的反应环境. 水环境中的PFOS和PFOA首先在空化气泡表面发生扩散和吸附,之后再在空化气泡和溶液之间的界面区域发生热解. 以PFOS为例,热解的具体过程是C—S键发生裂解,脱去SO3形成全氟烯烃中间体(CnF2n•),继而进一步热解成含有一个C的氟自由基(:CF2、•CF3),最后氟自由基与溶液中的•OH和H•发生氧化还原反应,最终变为HF、CO和CO2,其具体过程如图3所示[52].
声化学过程中超声频率的高低直接影响PFAS的降解效率,增加超声频率会加快产生空化气泡和促进水热解产生•OH[54]. 此外,超声模式也会影响PFAS的降解效率,Campbell等[55]研究了单频超声和多频超声对PFAS降解效果的影响. 以AFFF的降解为例,在使用双频的情况下,声波的重叠会产生非正弦的超声波形式,从而增强气泡膨胀以及缩短坍塌时间,提高空化效应并提高声化学反应效率.
虽然声化学方法具有较高的降解效率,但超声过程中需要外部输入较大的能量,而能量的利用效率低。目前将超声与其他技术进行联用的方法,可以在提高声化学处理效果的同时,降低反应总能耗(表4). 比如在超声和微波联用的情况下,超声波的空化作用减少了等离子体产生时所需的微波功率,水中产生的等离子体促进了•OH、H•、HO2•和O1•自由基等活性物种的生成(式19)[56],并促进PFOA的等离子体热解分解. 此外,向声化学过程中添加化学试剂(过硫酸盐[57]和碳酸盐[58]等)也可以提高降解效率,加入过硫酸盐后,超声辐照引起的瞬间高温导致S2O82−热解形成SO4•−,促进PFOA的脱羧反应;而将碳酸氢钠加入声化学体系后,水热解产生的•OH会与HCO3−发生单电子氧化(式20),生成具有强氧化性的二次自由基CO3•−,能更好地诱导PFAS的降解(式21、式22)并减少污染物的持久性,最终热解为F−、CO和CO2[52].
声化学方法利用空化现象在温和条件下实现PFAS的高效降解,同时也能在较短时间内完成PFAS的彻底矿化. 与电化学氧化法相比,声化学需要更多外部能量的输入,超声过程中使用的气氛类型[61]、超声频率和功率密度等也会对降解效率产生很大影响. 针对声化学过程能耗大的问题,可以从以下几个方面提升去除效率:(1)改进声化学使用装置,优化超声种类,增强空化效应,提高反应速率;(2)耦合超声或其他高级氧化降解方法(光催化),加速PFAS脱氟过程的进行,减少运行能量消耗. (3)加入过硫酸盐、碳酸盐等化学氧化剂,产生额外的自由基从而促进降解反应的进行,减少总输入能量并进一步提高声化学处理的效果. 除此之外,表面活性剂的加入也会改变水相的表面张力、离子强度以及空化气泡界面区域的PFAS浓度,影响声化学的降解效率.
-
光催化法可以在温和、低能耗条件下降解污染物[62],大量研究表明了光催化处理PFCAs有着高的降解效率与脱氟率,是一种具有较好前景的水处理技术[63]. TiO2基催化剂[64 − 65]、杂多酸[66]、InOOH基催化剂[67]等都能被用作降解PFCAs,表5总结了不同光催化剂对PFOA的降解效果. 在光催化降解PFCAs过程中,主要是PFCA与活性物种之间发生了电子转移,光催化降解机理可以总结为两类:(1)光生电子-空穴对(e−-h+)氧化PFCAs;(2)杂多酸直接电子转移氧化PFCAs.
光生电子-空穴对直接氧化是光催化中的最常见的机理. 以最常见的半导体光催化剂TiO2为例,如图4所示,在UV光照下,TiO2会产生光生电子(e−)和光生空穴(h+)(式23),PFCA与h+之间发生电子转移形成PFCA自由基(式24),PFCA自由基自分解为CO2与CnF2n+1自由基(式25),下一步CnF2n+1自由基会转变为全氟醇,这一步在文献上有着不同的解释,有研究认为是H2O与h+发生电子转移生成•OH与CnF2n+1自由基结合[65](式26),也有研究认为是O2与H2O的共同作用[63](式27—29),全氟醇是一种不稳定的化合物可以分解成为酰基氟类化合物(式31),酰基氟进一步水解生成少一个C的PFCA(式32),这样的反应不断循环就可以将PFCA矿化。除此之外,式28形成的CnF2n+1O•还可以发生β裂解,生成Cn-1F2n-1•与COF2(式30).
对基于光生电子-空穴对的降解机理来说,如何延长光生电子-空穴对的寿命是非常重要的. 在单独的TiO2体系中,PFCA与h+发生电子转移后,电子会积累在TiO2的导带上导致光生电子与光生空穴重新结合的速率加快,光催化效果下降[63],因此提高光催化剂光生电子-空穴对的分离效率最重要. Chen等[65]在TiO2上掺杂过渡金属(Fe、Cu)对PFOA进行降解,研究表明掺杂过渡金属的TiO2催化剂相比于单独的TiO2有着更高的降解效率与脱氟率,使用UV/Cu-TiO2体系降解初始浓度为50 mg·L−1的PFOA,反应12 h有着91%的降解效率与19%的脱氟率,PFOA的一级降解反应速率常数是UV/TiO2体系的30倍,效率的提高可以归结于过渡金属产生电子阱捕获光生电子,提高了光生电子-空穴对的分离效率. 除此之外还可以利用光生空穴的氧化能力去得到自由基来辅助降解PFCA,Wang等[64]利用草酸与光生空穴之间的电子转移,得到具有强还原能力(E0(CO2/ CO2•−)= −1.85 V)的羧酸阴离子自由基(CO2•−),UV/H2C2O4-TiO2-N2体系降解PFOA的效率比UV/TiO2-N2体系提高了7倍.
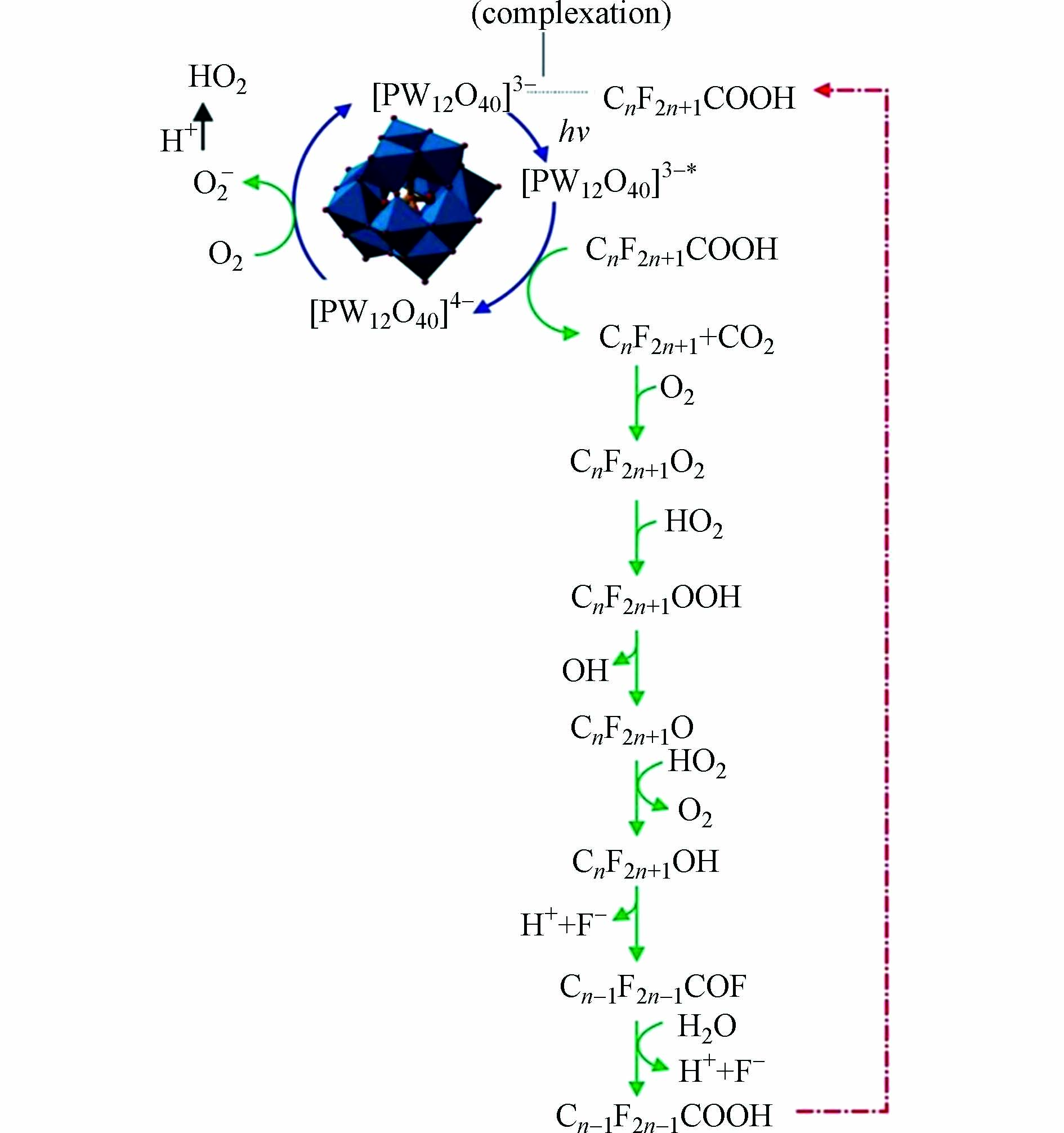
杂多酸也可以有效地降解PFCAs. You等[62]使用磷钨酸(H3PW12O40,HPW)负载在双峰介孔二氧化硅(BMS)去降解初始浓度为5 mg·L−1的PFOA,30 %wt HPW-BMS-VUV降解PFOA效果最好,4 h后脱氟率可达53%. 杂多酸催化降解PFCAs主要依靠杂多酸阴离子激发态的强氧化能力,以HPW为例解释直接电子转移的降解机理,如图5所示. 在UV光下,PW12O403−由基态跃迁为激发态PW12O403−*(式33),激发态的PW12O403−*与PFCA发生电子转移形成PW12O404−与CnF2n+1COOH+(式34),CnF2n+1COOH+的C—C键裂解得到CnF2n+1•(式35),在氧气的存在下PW12O404−被氧化为PW12O403−(式36),同时CnF2n+1•也可以和O2反应得到CnF2n+1OO•(式27),继而裂解形成CnF2n+1O•(式28). CnF2n+1O•将会有两种途径的转化[62],一种是自分解形成Cn−1F2n−1•与COF2(式30),另一种是与HO2形成全氟醇(式37),其中HO2可由O2−与H+结合而得(式38),生成的全氟醇将会按照式31、式32进行下一步反应. 体系中杂多酸阴离子可以不断地被激发、还原、氧化,促使了PFCA可以不断地进行降解.
光催化法虽然能够有效地降解PFCAs,且具有不需要额外添加化学试剂、条件温和、操作简便等优点,但是却受到诸多条件的限制,影响了其对于实际体系的应用. 对于传统的光生电子-空穴对的光催化来说,提高光生电子-空穴对的分离效率是最为重要的,可以通过掺杂过渡金属去捕获光生电子,从而提高光生电子-空穴对的寿命;对于新型杂多酸催化来说,主要利用了阴离子激发后的强氧化能力,因此需要阴离子能够在溶液中稳定存在,但是对于HPW来说,其阴离子在pH=1左右才能够稳定[66],因此需要发展新型的杂多酸催化剂使得其阴离子能在更宽的pH范围稳定,从而拓宽杂多酸的使用范围,此外水溶性的杂多酸也很难从溶液中回收,会影响后续水体处理,解决杂多酸的回收也是必要的. 为了促进光催化的实际应用,以下几个问题值得进一步探究:(1)延长光生电子-空穴对的寿命;(2)提高光催化剂比表面积,使其具有更多的活性位点;(3)回收水溶性的杂多酸催化剂,重复利用降低成本;(4)通过掺杂技术减少半导体光催化剂带隙,从而拓宽光吸收范围;(5)提高光源覆盖性,从而提高光处理规模.
-
基于水合电子(eaq−)的高级还原技术(advanced reduction processes,ARPs)是高效降解水环境中PFAS的重要手段之一[77 − 78]. eaq−具有活性高、还原性强的特性,是一种强的还原物种(E=−2.9 V,t1/2>300
$ \mathrm{\mu }\mathrm{s} $ ),可以与有机物中的卤素原子发生反应,在降解PFAS过程中通过破坏C—F键以达到高效脱氟和降解的效果[77 − 79]. eaq−的产生方法有很多,光化学中通常使用紫外光活化H2O、亚硫酸盐(UV/S)[80 − 82]和碘化物(UV/I)[79, 83 − 84]等试剂[78]来获得,其机理如式(39—42)所示,式中($ {\mathrm{I}}^{\cdot},{\mathrm{e}}^{-} $ )表示I•与eaq−成对形成溶剂笼,*表示I•H2O−处于激发态[78]. 这一章节主要综述了eaq−与全氟类化合物之间的反应,如PFCAs与PFSAs的降解机理. -
eaq−降解PFCAs有着优秀的降解率和脱氟率,如表6所示,展示了不同方法产生水合电子对PFCAs的去除效果. 使用UV/S体系降解不同链长初始浓度为25 μmol·L−1的PFCAs,8—12 h内可达到近100%的降解率,在48 h左右获得约55%的脱氟率,三氟乙酸(TFA)脱氟率甚至可以达到100%[85];使用UV/I体系降解初始浓度为0.025 mmol·L−1的PFOA,降解6 h后降解率和脱氟率分别为93.9%、76.8%[79];近期研究发现,相较于UV/S体系,UVC/S+I复合体系降解PFCAs在脱氟率与降解率上均有1—2倍的提高,同时也能降低反应能耗[84],是一种经济、高效的新方法.
目前来说,eaq−降解PFCAs的机理主要分为两类:(1)H/F交换机理;(2)链缩短机理,图6展示了PFCAs被水合电子进攻的两种不同路径. 此外,在最新研究中Ren[77]等首次提出了在UV/S体系降解PFOA过程中还存在着SO3•−/F交换的机理.
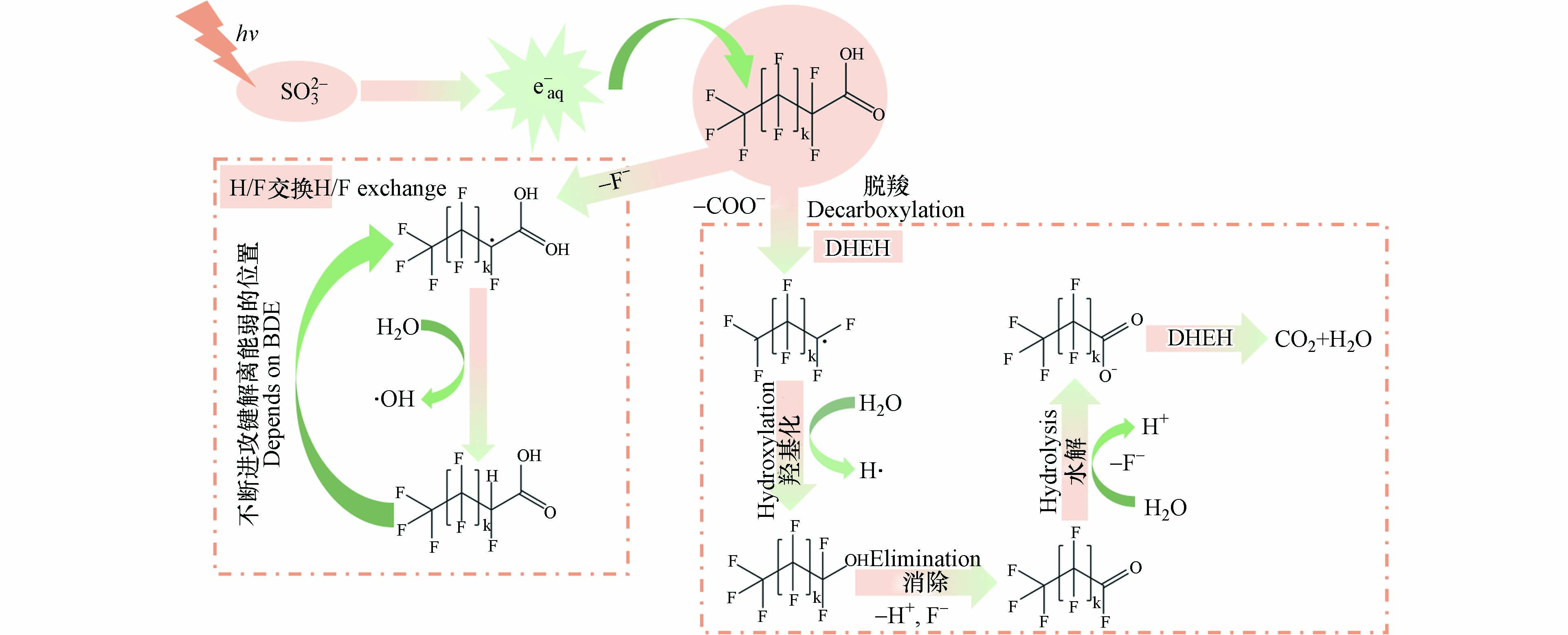
在PFCAs的H/F交换降解途径中,eaq−作为亲核试剂可进攻与—COOH相邻的α位上的C—F键形成•CnF2nCOO−(式43),这是因为在Bental等[89]研究中,密度泛函理论(DFT)计算得到α位上的C—F键的键解离能(BDE)比碳链上其他位置的C—F键的键解离能低. 生成的产物•CnF2nCOO−和H2O发生H的加成完成H/F交换得到CnF2nHCOO−(式44),根据C—F的BDE的不同,eaq−会不断进攻BDE低的C—F键,最终得到多氟烷基羧酸(式45).
在PFCAs的链缩短机理中存在着两种解释:(1)H/F交换直接光解;(2)脱羧-羟基化-消除-水解(DHEH). 对于第一种链缩短途径来说,经历两次H/F交换后的产物CnF2n-1H2COO−在UV照射下可分解为•Cn-1F2n-1、•COO−和:CH2(式46),产物•Cn-1F2n-1与•COO−将会结合形成新化合物Cn-1F2n-1COO−(式47),Cn-1F2n-1COO−会继续重复上述过程,通过两次H/F交换与直接光解,不断脱去:CH2,从而缩短碳链最终矿化. 但是在H/F交换直接光解途径中存在着两个问题:(1)—CH2—的存在会导致PFCA更难降解,在Bental等[85]实验中发现当—CH2CH2—基团插入PFCA后,其脱氟率由56.5%下降至17.1%(n=7,PFOA),因此两次H/F交换的直接光解过程很难发生(式46);(2)在溶液中•Cn-1F2n-1与•COO−的浓度很低,两者结合的可能性小[78](式47),影响了后续进行两次H/F交换. 因此,大多数研究使用DHEH机理去解释PFCAs的链缩短过程.
在DHEH机理中,首先是eaq−进攻PFCA与—COOH相接的C—C键(也可以直接在UV光照下裂解),eaq−破坏掉α位的C—C键形成•CnF2n+1和COO−(式48),接着•CnF2n+1与H2O发生羟基化得到羟基化产物CnF2n+1OH(式49),CnF2n+1OH进行F−的去除得到Cn-1F2n-1COF(式31),Cn-1F2n-1COF通过水解反应得到Cn-1F2n-1COO−(式32),Cn-1F2n-1COO−又可以进行DHEH反应,最终PFCA被完全矿化为CO2(式50). 在DHEH机理中,值得注意的是脱羧中对C—C键的进攻过程,因为直接光照裂解C—C键是没有证据支撑的假设,而且eaq−对C—C键的裂解在现有文献中也没有阐述清楚,eaq−驱动的C—C键裂解的假设是根据低能电子可以诱导凝聚相的甲酸和TFA脱羧提出的[78]. 因此相较于第一种机理,使用DEHE机理去解释PFCAs的链缩短过程是更合理的.
-
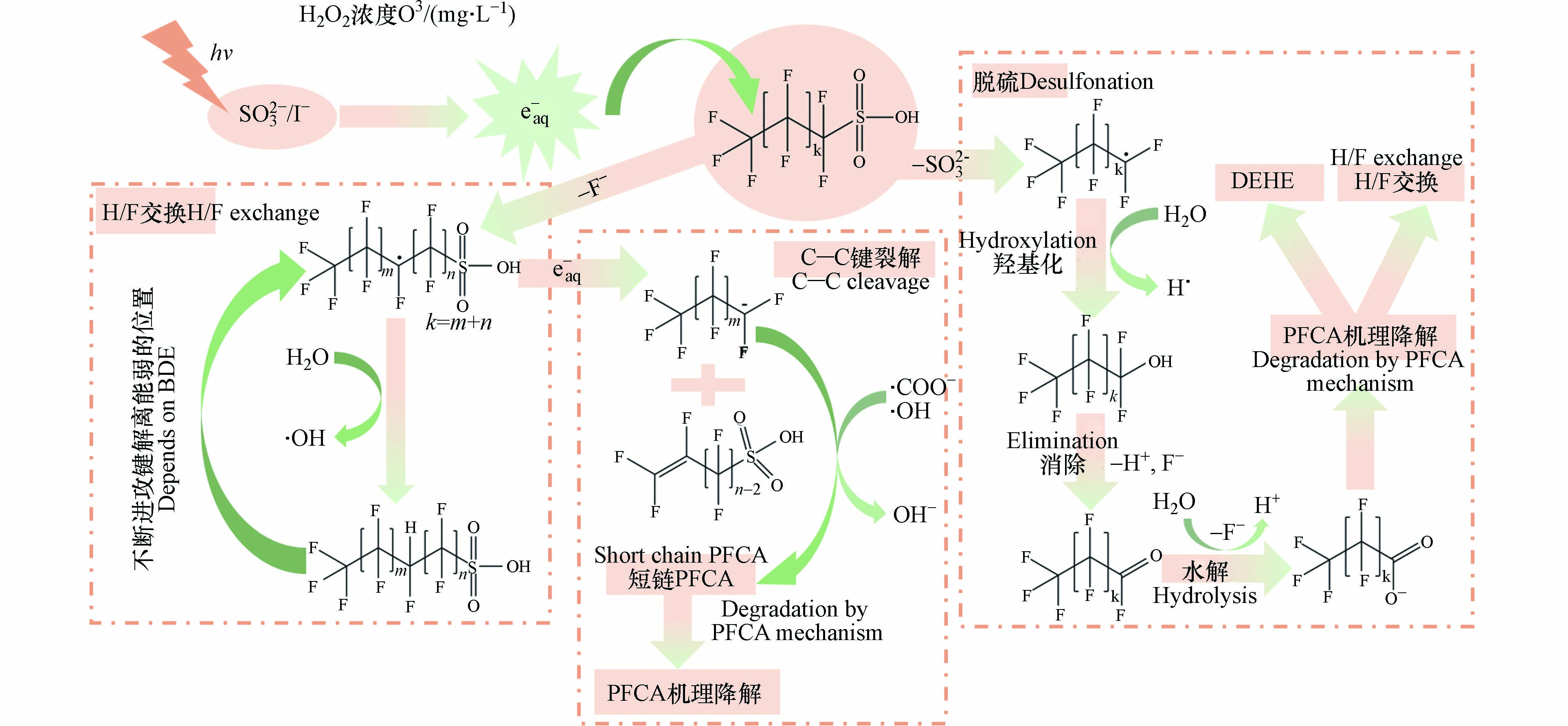
eaq−对PFSAs也拥有着优秀的降解率与脱氟率,如表7所示,展示了不同方法产生水合电子对PFSAs的去除效果. 例如,使用UV/S体系降解初始浓度为37.2 μmol·L−1的PFOS,4 h可以达到85.8%的降解率和64.6%的脱氟率[80]. 对于PFSAs的降解过程来说,其降解机理分为3类:(1)H/F交换;(2)脱硫;(3)C—C键裂解,图7展示了3种不同的降解途径.
PFSAs的H/F交换机理与PFCAs的H/F交换机理相同,都是eaq−去进攻C—F键,但是eaq−攻击的位置不同,PFSAs中eaq−更喜欢进攻氟烷基链上中间的C—F键[78]. 因为—SO3H代替了PFCA中的—COOH基团,导致了PFSA与PFCA的结构性质发生改变,氟烷基上不同位置的C—F键BDE也受到影响. 比如在烷基链n=4的PFSA上,DFT计算得到β位上的C—F键BDE(445.6 kJ·mol−1)比α位上的C—F键BDE(456.9 kJ·mol−1)低,所以eaq−会优先进攻β位的C—F键[85]. 总的来说,eaq−进攻时会优先进攻C—F键BDE更低的位置,随之在进攻位置发生H的加成完成H/F交换.
在脱硫机理中,由于PFSA中C—S键的键能(272 kJ·mol−1)比PFCA连接头部基团的C—C键的键能(346 kJ·mol−1)低,使得C—S键容易断裂[78]. eaq−进攻PFSA后,会导致C—S键的拉长[85],最终结果是C—S键的断裂(式51、式52)形成•CnF2n+1,在碱性条件下会得到少一个C的PFCA(式49、式31、式32),PFCA可以按照上述提到的DHEH机理或者H/F机理进行脱氟与降解.
PFSAs的C—C键裂解机理是基于前线轨道理论提出的,eaq−会加成在PFSAs的最低电子未占据分子轨道(LUMOs)上,导致C—C键键级变小,C—C键作用力被削弱而发生裂解. 上述过程可以有两种途径[78],一种是eaq−加成在烷基链上中间的—CF2—,然后与溶液中的H+进行反应从而裂解C—C键;另一种是首先进行F−的脱除,然后eaq−与负离子自由基进行加成,随之发生C—C键的裂解. 但是对于ARPs过程来说,eaq−在碱性下更易存在,在酸性条件下会被H+猝灭,因此第二种裂解途径更为合理,其具体反应如式(53)—式(55)所示. PFSA在经过F消除后会得到•CnF2nSO3−(式53),eaq−加成后形成负离子CnF2nSO32−(式54),CnF2nSO32−会发生解离导致C—C键的裂解(式55)[78]. 对于最为常见的PFOS来说,LUMOs存在于C4—C8原子上,C—C键的裂解会形成C3F7−、C4F9−、C5F11−及各类烯烃的中间体,这些中间体会与•COO−或者•OH形成PFCA,然后按照PFCA的降解模式进行脱氟与降解[78].
-
由于eaq−的强还原性,对PFAS有着较高的脱氟率和降解率,而且eaq−还能多途径的对PFAS进攻,利用多种进攻方式协同去除PFAS. 但是eaq−多种途径的进攻方式却带来了复杂的中间体,而且这些含氟中间体也具有一定的毒性. 对于H/F交换途径来说,会产生含F数量不一的多氟化合物;对于链缩短途径来说,会产生链长不同的短链全氟化合物;对于C—C键裂解途径来说,会产生含氟的烯烃化合物与全氟离子; eaq−的产生方式也会导致形成不同产物,多种中间体构成的复杂水体会对分析带来麻烦. 此外eaq−的活性也受到许多外部条件的影响,比如 pH过低(<7)、干扰离子(NO3−)、溶解氧的存在会对eaq−产生猝灭作用. 另外,高级还原法需要额外添加化学试剂去产生eaq−,残留的化学试剂也会对水体造成二次污染. 常用的UV/S和UV/I体系中残留的亚硫酸盐和碘化物都对人体具有毒性,UV/3-吲哚-乙酸毒性低且不受pH限制的体系,但是其与PFAS的反应速率缓慢,很难应用于实际体系[82]. 为了促进水合电子还原法在PFAS去除中的实际应用,更多的研究可以关注在:(1)处理PFAS前先对溶液进行预处理,可以通过离子交换树脂与通入氮气去除水中对水合电子干扰的离子与氧气;(2)提高水合电子产量,从而对中间产物进行深度矿化;(3)探究新体系产生水合电子,避免残留试剂对水体的二次污染.
-
微生物降解技术是一种经济又环保的方法,但是PFAS中的C—F键能高达536 kJ·mol−1,生物降解PFAS的反应周期长. 通过DFT理论计算发现,微生物还原脱氟是可行的[95 − 96],但是目前对PFOA和PFOS的生物降解特别是矿化的研究非常有限.
早在2010年,Liou等[97]研究PFOA的生物降解性能,研究团队选用了5种不同的生物菌落在厌氧条件下,分别用醋酸盐、乳酸盐、乙醇和氢气作为电子供体,PFOA作为电子接受体进行培养,测试了在4个厌氧终端电子接受过程(TEAPs)中PFOA的共代谢数据,因质谱分析没有获得PFOA的转化产物,认为PFOA在微生物降解上是惰性的. 最近新兴起的嗜酸菌科菌株 A6被报道可以应用于降解 PFOA、PFOS. A6是一种自养生物,在PFOA、PFOS存在下,可氧化铵盐试剂NH4+成为NO2−、还原Fe3+成为Fe2+[95]. Huang等[95]使用A6去降解初始浓度为0.24 mmol·L−1的PFOA和0.20 mmol·L−1的PFOS,经过100 d的降解,发现高达60%的PFOA、PFOS被去除. 除了A6以外,Luo等[98]研究了PFOA在酶催化氧化腐殖酸反应中的分解情况,在添加漆酶和1-羟基苯并三唑的缓冲溶液中处理PFOA(初始浓度为1 μmol·L−1),反应157 d后 PFOA去除率和脱氟率分别为50%和28.2%. 微生物降解也可以和其他技术耦合使用提高PFAS的降解效率. Ding等[99]使用Bi12O17Cl2作为光催化剂协同活性污泥中的微生物群落降解初始浓度为500 μg·L−1的PFOA,700 min达到了79.7%的去除率,总有机碳(TOC)去除率和脱氟率分别达到57.4%和57.6%.
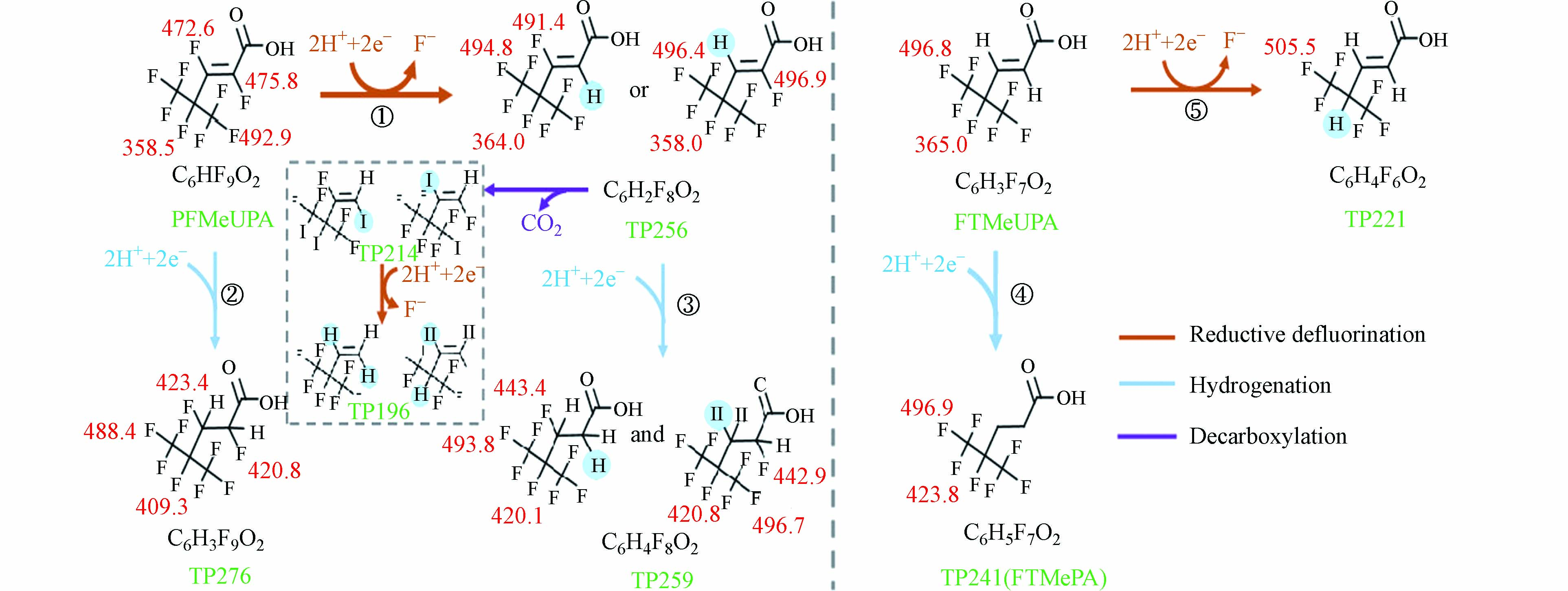
对于不饱和的PFAS来说,更适合使用微生物法进行降解,Yu等[96]研究了两种不饱和的全氟或多氟化合物(PFMeUPA和FTMeUPA,初始浓度约75 μmol·L−1),在有机卤化物呼吸微生物群落中分别处理FTMeUPA 70 d和PFMeUPA 130 d,FTMeUPA与PFMeUPA去除率都达到>90%,脱氟率约为20%和75%. 微生物法降解不饱和含氟化合物,主要有两种途径,如图8所示:(1)还原脱氟. 在微生物的作用下,首先断裂sp2上的C—F键 . 对于没有sp2 C—F键的多氟化合物,微生物作用下则是会先进攻BDE小的sp3 C—F键;(2)加氢反应. 微生物的作用下,C=C双键发生加成形成相应的饱和PFAS[96]. 2022年Yu等[100]进一步研究了不饱和含氟化合物(FCAs)脱氟与结构之间的关系(图9),研究表明α,β不饱和结构是厌氧微生物脱氟的关键,不饱和全氟化合物更优先发生还原脱氟而不是加氢,而没有sp2 C—F键的FCAs在厌氧条件下加氢比脱氟更有利.
总体来说,微生物降解PFAS法成本低且绿色环保,但是降解周期长、降解难度大. 不饱和PFAS更容易受到微生物的青睐,因此,使用微生物处理不饱和的PFAS是更有优势的;对于饱和PFAS来说,使用微生物耦合其他方法(比如高级氧化法或者高级还原法)会更有效率. 因此如何将微生物与其他去除技术结合是未来发展的方向,不仅能提高去除效率,而且还能降低成本. 比如说,将高级还原法与微生物降解法进行结合,还原法在处理PFOS时,C—C键裂解会产生复杂的不饱和PFAS,再通过微生物和水合电子的共同作用,对还原法中的复杂中间产物进行共处理,有效降低复杂中间产物带来的二次毒性.
-
表8为不同方法去除PFAS的优缺点对比.在高级氧化法中,过硫酸盐活化和电化学的方法会产生大量短链的PFAS,短时间内无法实现彻底矿化. 反应条件(例如温度、pH等)对两种方法降解污染物效率也有影响. 对于活化过硫酸盐体系,在适当的温度范围内,温度的升高会促进SO4•−的产生;而当温度进一步升高时,反而会发生SO4•−的清除反应,影响降解效率. 在电化学反应中,对反应体系进行水热时温度的升高会增强电极和溶液的电化学性能,并促进电极表面•OH的形成. 除污染物种类、污染物浓度、电流密度等基本参数,电极材料的电导率、析氧电位等电化学性能也会影响PFAS降解效果. 活化过硫酸盐和电化学的方法尽管可以实现PFAS高去除率,但在反应过程中短链PFAS的产生从一定程度上限制了其在实际水体中的运用. 电化学和活化过硫酸盐法的降解效率易受到实际水体中杂质离子的影响,目前并未扩展到实际水体的原位处理中. 声化学方法能在较短的时间内将PFAS降解且不需要加入催化剂,但其降解性能高度依赖反应温度、频率和功率密度等参数,最近的研究更偏向于对声化学参数的优化和装置的改进. 声化学处理技术虽降解效率快,实际使用时需要大量的能量输入,经济成本高,难以实现大规模的应用. 光催化剂的电子-空穴对分离效率低是目前光化学还原法存在的主要问题,可以通过掺杂过渡金属形成电子阱捕获光生电子,从而提高电子-空穴对分离效率. 对于杂多酸来说,受到pH、回收等限制,拓宽pH应用范围和回收杂多酸降低成本是比较重要的.
在高级还原法中,主要以光化学产生水合电子作为还原剂,水合电子可以多种进攻方式协同去除PFAS,实现高的脱氟率和降解率,但在降解的过程中会产生具有一定毒性的复杂中间体. 水合电子pH受限也会导致使用范围较窄. 此外,反应过程中,溶液pH值、反应温度、水溶液中的共存离子、溶解氧等外界因素也会对水合电子的活性产生影响,水合电子的产生需要加入额外的化学试剂,易产生二次污染. 光化学还原法需要额外引入紫外光,目前还只能处理小规模废水,大规模废水的应用仍需进一步探究.
微生物降解法的成本较低且绿色环保,并且在处理不饱和PFAS时更具有优势. 但由于其代谢周期长、代谢效果不佳、易受降解污染物种类影响和产生有毒中间产物等因素导致其目前阶段难以运用于实际水体的处理.
-
PFAS类化合物在食品包装、服装、家具等行业中的广泛使用使其遍布全球各地,PFAS稳定的化学结构使其具有难降解性和高生物毒性,很难在自然环境下降解,是目前广受关注的新污染物. PFAS可以通过多种环境介质实现长距离的迁移,尤其是通过食物链传播在生物体内富集,对人类健康等产生潜在威胁. 高级氧化法、高级还原法和微生物降解法目前已被广泛应用于PFAS的去除. 在PFAS的降解过程中,无论是哪种方法,都不可避免地会产生大量的短链氟化物,而这些短链的氟化物与长链的氟化物具有相似的结构和化学性质,因此中间体的生物毒性、脱氟率和TOC去除率是评价降解方法的主要依据. 随着PFAS在土壤和水体中的被频繁检测出,传统的处理技术还需要进一步改进,特别是使其要满足目前不断提高的水处理要求,同时也要考察处理技术在实际水环境中的适用性. 对PFAS治理方法的探索,不仅是降低环境基质中PFAS对环境和生物体的危害,更是对实现经济的可持续发展和社会的和谐发展具有重大的理论和实践意义. 经济的发展依赖于环境和资源,而在经济高速发展的同时也往往也伴随着环境污染问题的出现. 正确处理好环境与经济的相互关系,对环境进行治理和保护,才能更好的进行可持续性发展,实现人与自然的和谐共处.
随着PFAS的关注越来越多,其去除研究也正在不断地进行更新,除了上述所提到高级氧化法、高级还原法以及微生物法外,还发展了其他的新方法. Zhao等[101]研究了非热等离子体(介质阻挡放电等离子体)技术对于PFOA的去除,初始浓度300 mg·kg−1,在反应2 h后可以达到大于80%的去除率;2022年PFOA的去除登上了《科学》杂志,Trang等[102]在80—120 ℃和环境压力下,在二甲基亚砜(DMSO)和水混合物中加入NaOH进行PFOA的矿化,其脱氟率>90%.
在未来处理PFAS的研究中,光电处理是值得我们去进一步研究的. 尽管目前对PFAS去除的研究较多,但都还需要进一步的完善. 新型检测技术的开发利用也为深入探究PFAS的降解机理提供充足的理论基础和科学依据,以期实现PFAS的彻底矿化. 实际水体中环境影响因素 (共存离子、共存有机物、pH值、水质背景等) 对降解方法的影响也需要进一步研究,并给出可量化的影响指标,才能更好的拓展在实际应用范围. 除此之外,对现有去除技术的改进和联用也将是PFAS去除领域的一个长期热点. 在之后的研究中,电化学氧化工作的重点应设计和构筑电化学活性高的电极材料,以及探究如何将多种电极材料复合达到更加优异的降解性能. 在光化学中,则是提高电子-空穴对分离效率、扩大光处理反应器,研究对实际废水的处理效果. 此外,多种处理技术的耦合也将是未来的发展趋势. 光电化学、热电化学、电凝技术等新颖方法,均体现出高去除效率和低能耗的优势.
全氟和多氟烷基化合物去除进展
Advances in removal of per- and polyfluoroalkyl substances
-
摘要: 全氟和多氟烷基化合物 (per- and polyfluoroalkyl substances,PFAS) 是指碳链上的氢原子被部分或者全部取代为氟原子的一类人工合成的有机化合物,因具有优异的疏水性、疏油性、抗污性及化学稳定性,被广泛运用于化工、服装家具、食品包装和日常用品等行业中. PFAS作为一种持久性有机物,会使人体免疫系统受损、甲状腺激素分泌紊乱甚至是引发癌症,而且PFAS性质稳定可长距离迁移,具有难降解性和生物蓄积性,是目前最受关注的新污染物之一,因此探索高效、节能的PFAS的去除方法具有重要的现实意义,也是环境科学领域的重要研究内容. 在全氟和多氟类化合物的降解中,主要利用了强氧化性物种(SO4•−、•OH和h+)或强还原性物种(eaq−)对PFAS进行进攻从而进一步实现裂解矿化. 本文综述了高级氧化法、高级还原法和微生物降解法这三种方法的技术特点,去除效果,反应途径和去除机理,并分析各方法的优点和不足之处,对全氟和多氟烷基化合物的去除新方法和发展前景提出展望.
-
关键词:
- 全氟和多氟烷基化合物 /
- 去除途径 /
- 高级氧化法 /
- 高级还原法 /
- 微生物降解.
Abstract: Per- and Polyfluoroalkyl substances (PFAS) are a class of synthetic organic chemicals in which one or more hydrogen atoms on the carbon chains are replaced by fluorine atoms. Owing to their exceptional hydrophobic, oil-phobic, stain resistant properties as well as their high stability, PFAS are extensively employed in a range of industries, including chemical, textile, furniture, food packaging and consumer goods. Furthermore, PFAS as persistent organic compounds have the potential to cause immune system damage, disruption of thyroid hormone secretion, and carcinogenic effects. Additionally, the stability of PFAS allows them to migrate over long distances, resulting in high levels of bioaccumulation and persistence in environment. PFAS have emerged as a significant concern among emerging contaminants, and there is an urgent need for efficient PFAS removal strategies. In the degradation of Per- and Polyfluoroalkyl substances, strong oxidizing species (SO4•−, •OH and h+) or strong reducing species (eaq−) are mainly used to attack PFAS and realize cleavage of C—F and C—C and further mineralization. This review provides a detailed discussion of three types of PFAS treatment processes, including advanced oxidation/reduction process and microbial degradation. The technical features, removal performance, removal pathway and removal mechanism are throughly summarized. In addition, the advantages and disadvantages of each method are highlighted. Through this review, we aim to draw attention to the research and the development of efficient technologies for PFAS removal. -

-
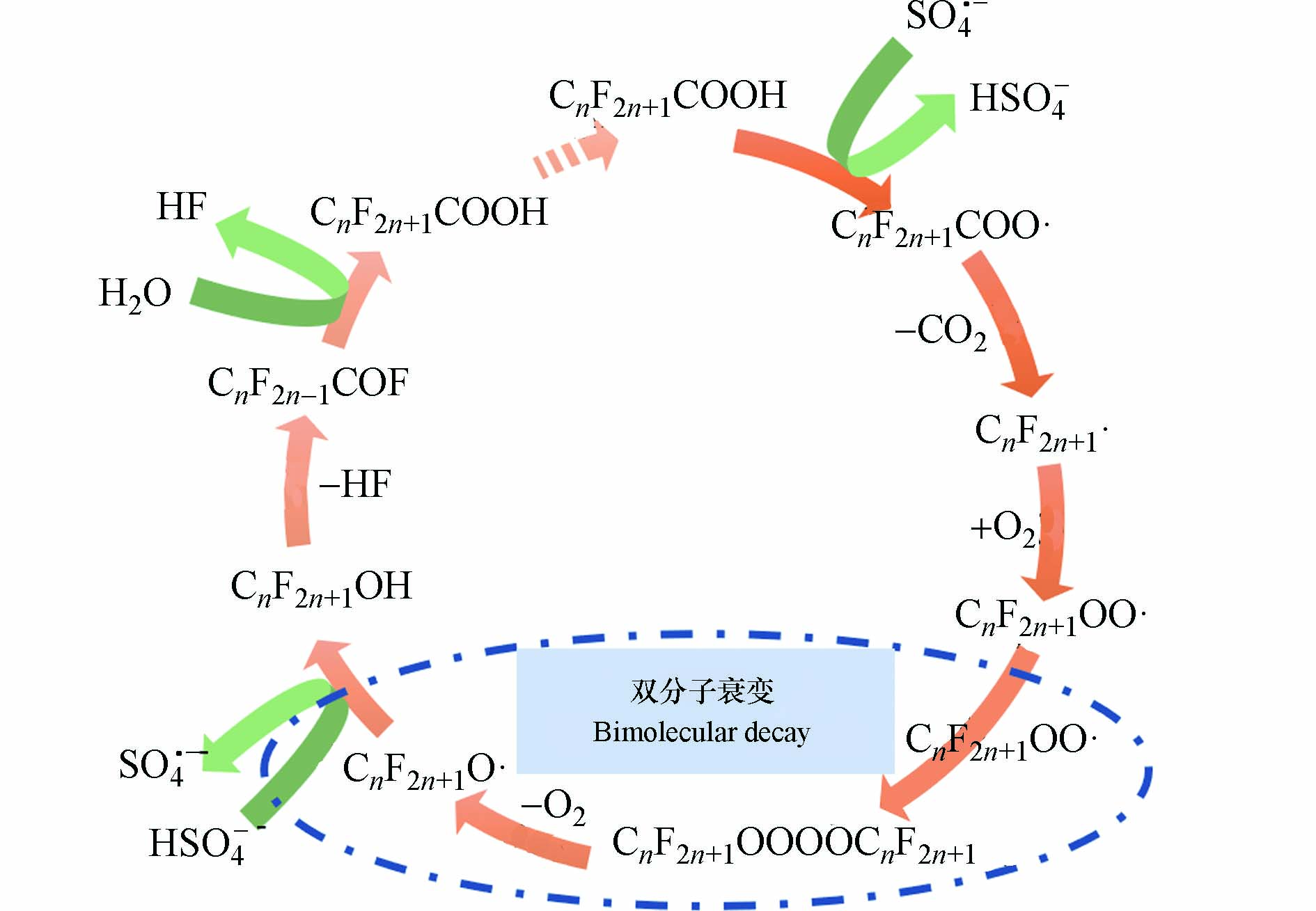

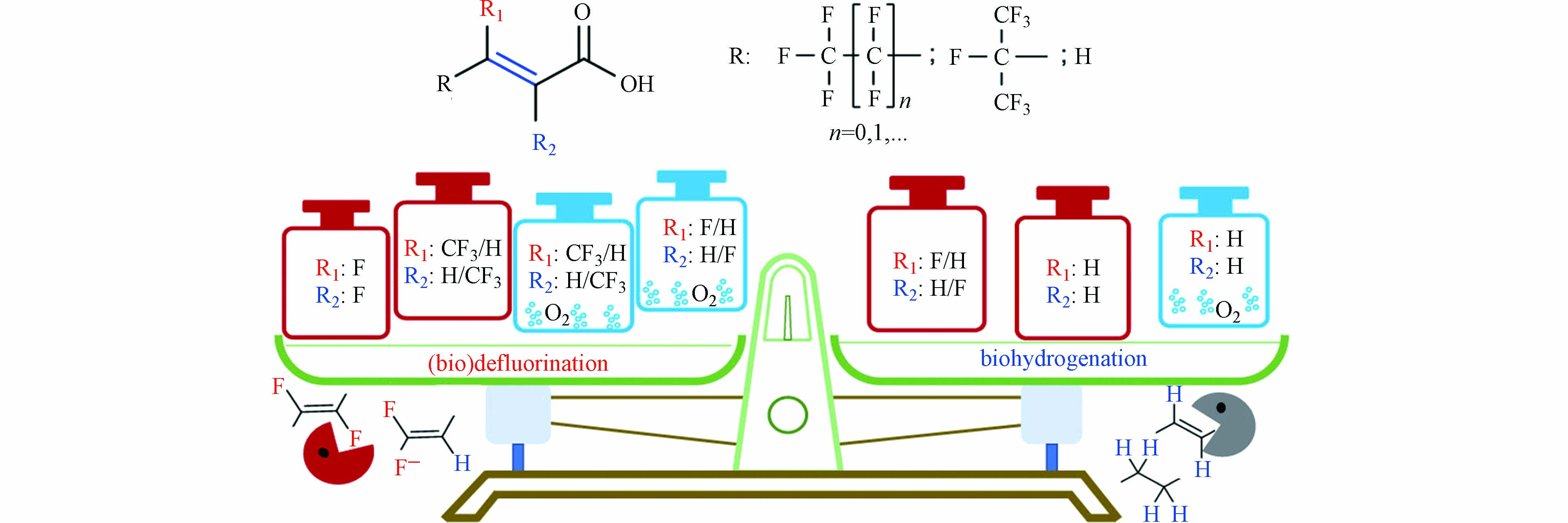
表 1 PFAS的结构以及物理化学性质
Table 1. The structure and physicochemical properties of PFAS
类别
Category名称
Compound
name分子式
Molecular
formula分子量/
(g·mol−1)
Molecular
weight结构式
Structure formula溶度积
ClogSa
Solubility
product密度/
(g·cm−3)b
Density沸点/
℃b
Boiling
point疏水常
数ClogPa
Hydrophobic
constant全氟烷基
羧酸类
(PFCAs)全氟辛酸
(Perfluorooctanoic acid,PFOA)C8HF15O2 414.07 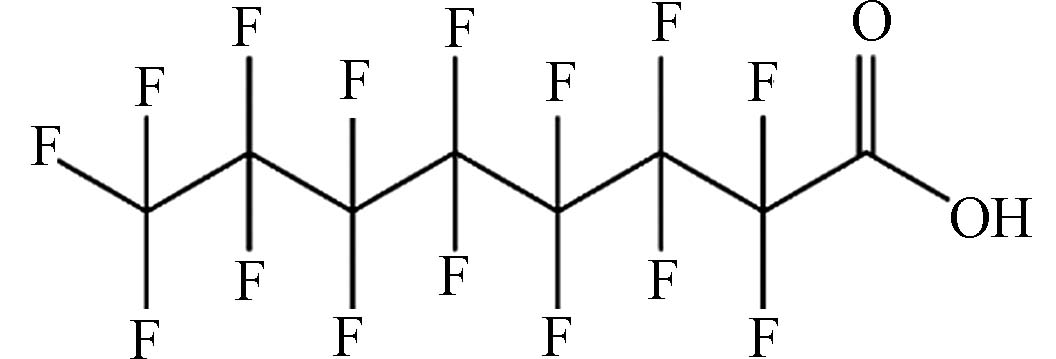
−5.507 1.8 189 4.232 全氟庚酸
(Perfluoroheptanoic acid,PFHpA)C7HF13O2 364.06 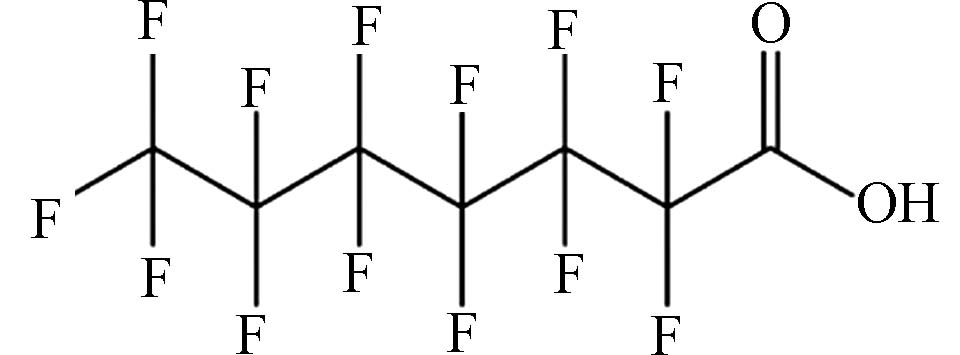
−4.789 1.792 175 3.611 全氟己酸
(Perfluorohexanoic acid,PFHxA)C6HF11O2 314.05 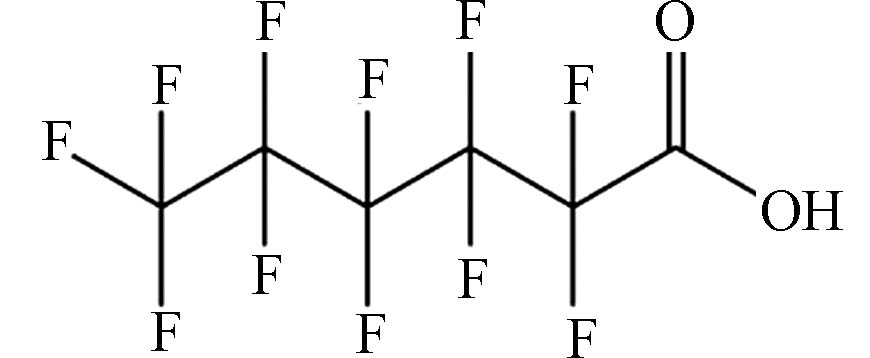
−4.070 1.759 157 2.99 全氟戊酸
(Perfluoropentanoic acid,PFPeA)C5HF9O2 264.05 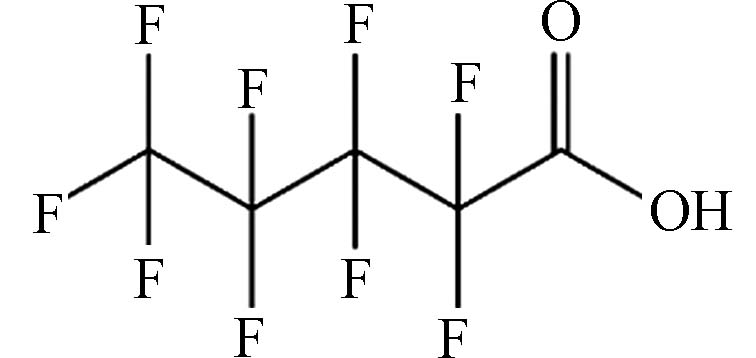
−3.352 1.700 140 2.369 全氟丁酸
(Perfluorobutanoic acid,PFBA)C4HF7O2 214.04 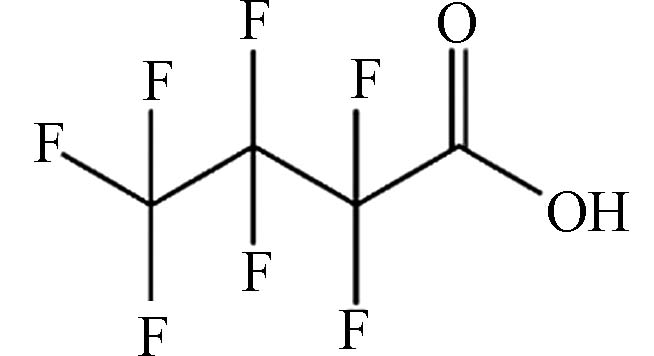
−2.635 1.645 120 1.748 全氟丙酸
(Perfluoropropionic acid,PFPrA)C3HF5O2 164.03 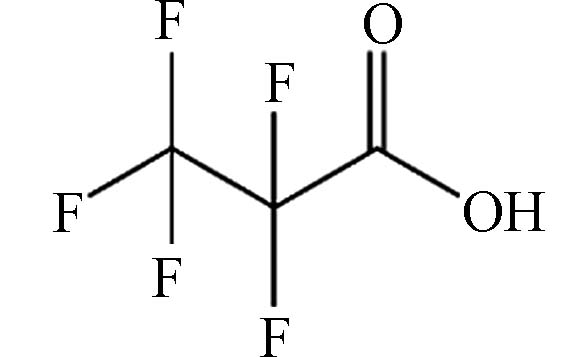
−1.916 1.561a 96-97 1.128 全氟烷基
磺酸类
(PFSAs)全氟辛烷磺酸
(Perfluorooctanesulfonic,PFOS)C8HF17O3S 500.13 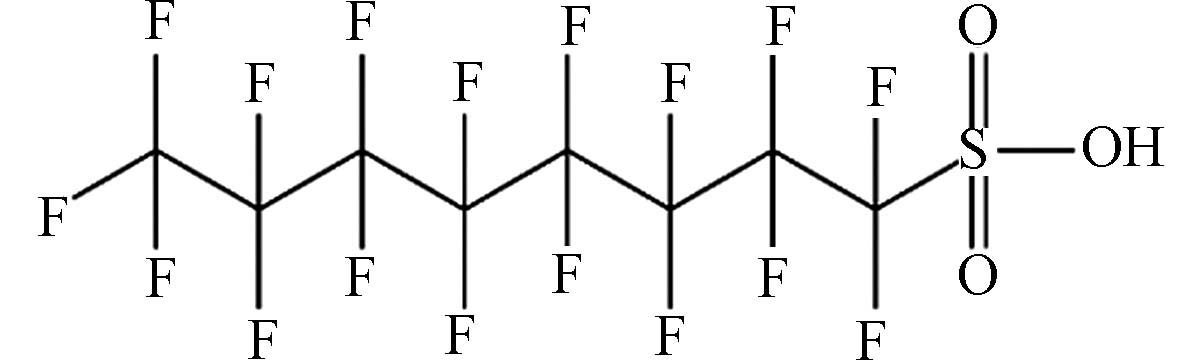
−6.254 1.25 260 3.461 全氟烷基
磺酸类
(PFSAs)全氟庚烷磺酸
(Perfluoroheptanesulfonic acid,PFHpS)C7HF15O3S 450.12 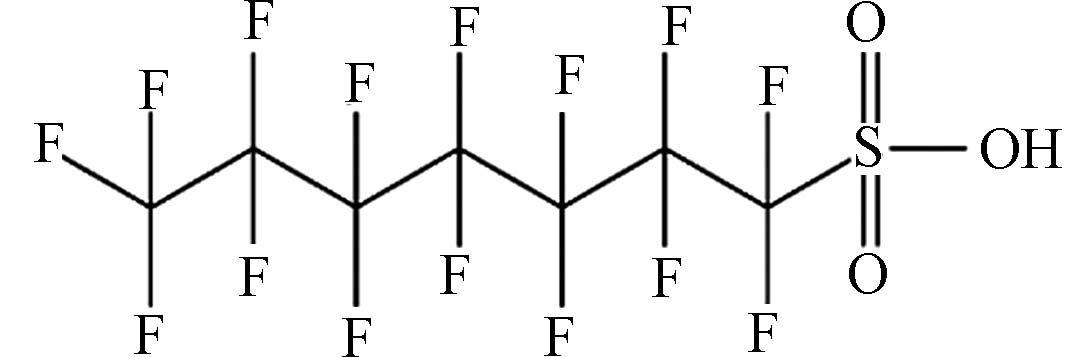
−5.536 — — 2.84 全氟己烷磺酸
(Perfluorohexanesulfonic acid,PFHxS)C6HF13O3S 400.11 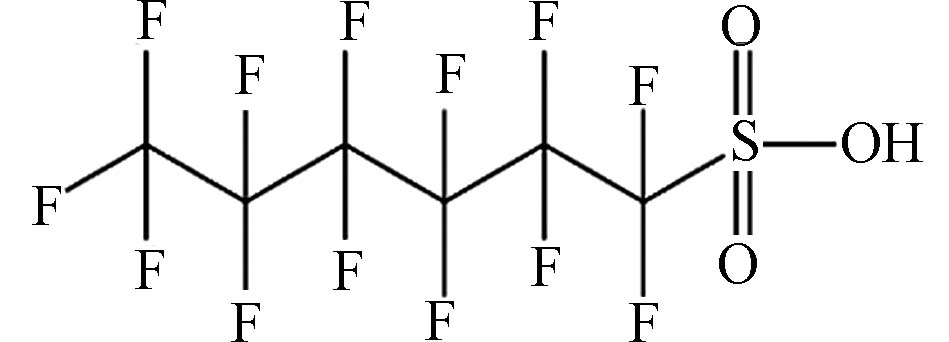
−4.818 1.841 238.5 2.219 PFAS
替代品六氟环氧丙烷二聚酸
(Hexafluoropropylene oxide dimer acid,GenX)C6HF11O3 330.05 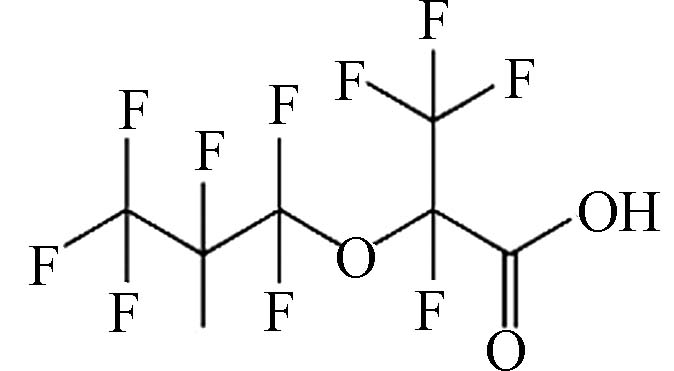
−3.885 1.748c 187.5c 3.1 6:2氟调磺酸
(6:2 fluorotelomer sulfonic acid,6:2 FTSA)C6F13CH2CH2SO3H 428.17 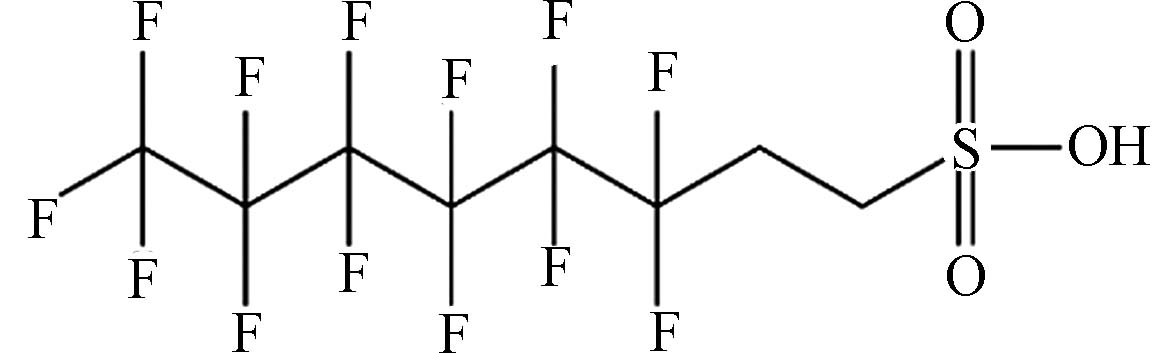
−4.808 1.953 — 2.767 6:2氟调羧酸
(6:2 fluorotelomer carboxylic acid,6:2 FTCA)C6F13CH2COOH 378.09 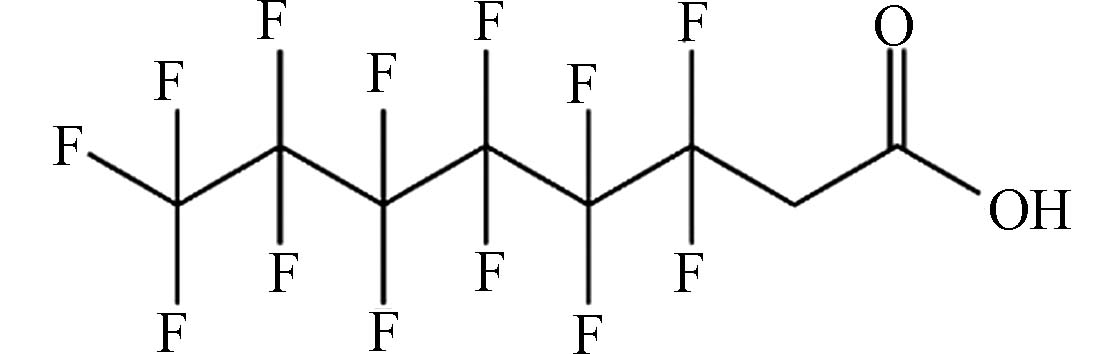
— — — — 6:2氟调醇
(6:2 Fluorotelomer alcohol,6:2 FTOH)C6F13CH2CH2OH 364.10 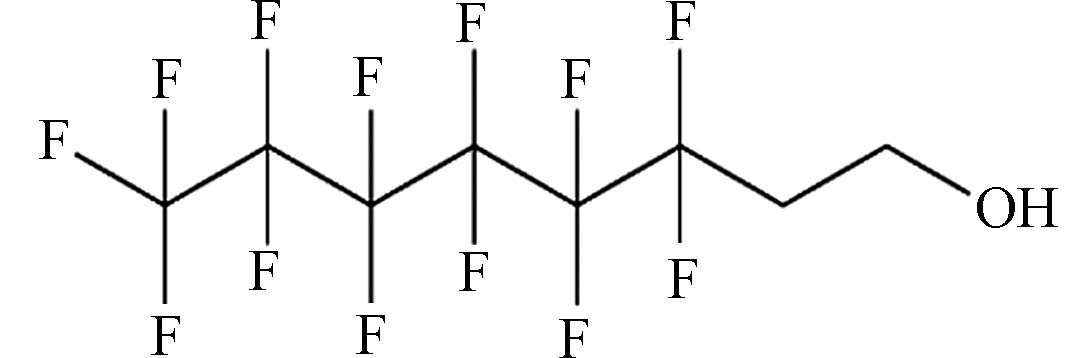
−5.076 1.651 88-95 4.475 注:a数据来源于Chemexper Chemical Director;b数据来源于化学品安全技术说明书(Sigma-Aldrich);c数据来源于化源网.
Note:a means data from Chemexper Chemical Director; b means data from Safety data sheet(Sigma-Aldrich);c means data from ChemSrc. 下载: 导出CSV
下载: 导出CSV
表 2 热活化过硫酸盐降解全氟和多氟烷基化合物的方法和效率总结
Table 2. Overview of PFAS degradation by thermal activation of persulfate
方法
Methods降解目标物
Degradation
target起始浓度/(µmol·L−1)
Initial concent-
ration实验条件
Experimental condition反应时间/h
Reaction
time降解效率/%
Degra-dation
rate参考文献
References热活化 PFOA 0.5 85 ℃,S2O82− 10 mmol·L−1,pH=7.1 30 93.5 [32] 热活化+Fe2+ PFOA 0.5 85 ℃,S2O82− 10 mmol·L−1,Fe2+
1.0 mmol·L−1,pH=7.130 80 [32] 热活化-微波 PFOA 253.8 90 ℃(70 W微波功率),S2O82−
5 mmol·L−1,pH=24 79.1 [28] 热活化-微波+Fe2+ PFOA 240.7 90 ℃(70 W微波功率),S2O82−
5 mmol·L−1,ZVI 3.6 mmol·L−11 58.5 [35]
下载: 导出CSV
表 3 不同阳极材料电化学性能对比
Table 3. Comparison of electrochemical properties of different anode materials
降解目标物
Degra-dation
target起始浓度/(mg·L−1)
Initial concent-
ration电流密度/(mA·cm−2)
Electric current
density阳极材料
Anode material电解质
Electrolyte反应时间/h
Reaction
time降解效率/%
Degradation
rate参考文献
ReferencesPFOS 0.6 15 BDD 10.5 mmol·L−1 Na2SO4 8 50 [49] PFOA 0.3 15 BDD 10.5 mmol·L−1 Na2SO4 8 90 [49] PFOA 100 20 Ce掺杂多孔纳米PbO2薄膜 10 mmol·L−1 NaClO4 1.5 96.7 [45] PFOS 46.5 30 Ti/TiO2-NTs/Ag2O/PbO2 100 mmol·L−1 Na2SO4 3 74.87 [50] PFOA 50 20 Ti/SnO2-Sb-Bi 12.3 mmol·L−1 NaClO4 2 99 [41] PFOS 100 100 F和Sb共掺杂Ti/SnO2 10 mmol·L−1 NaClO4 2 99 [44] PFOA 100 20 SnO2-Sb/CA 100 mmol·L−1 Na2SO4 5 91 [51] PFOA 100 20 SnO2-Sb/CA 10.5 mmol·L−1 Na2SO4 5 91 [51] PFOS 50 5 多孔Ti4O7陶瓷 20 mmol·L−1 NaClO4 3 93.1 [43] PFOS 1×10−4 10 Ag/Au-PAA/PAH 100 mmol·L−1 Na2SO4 2 91 [46] PFOA 1×10−4 10 Ag/Au-PAA/PAH 0.1 mol·L−1 Na2SO4 2 72 [46] PFAS 2.7×10−5 20 ZnO包覆的Ti 污水处理厂废水样 0.7 39—66 [47]
下载: 导出CSV
表 4 声化学降解方法及其降解性能总结
Table 4. Summary of sonochemical degradation methods
方法
Methods污染物
Contaminate起始浓度/(μmol·L−1)
Initial concentration超声频率及功率密度/功率
Ultrasonic frequency and
power density/power反应时间/h
Reaction time降解效率/%
Degradation rate参考文献
References超声 PFOA 20 200 kHz,8 W·cm−2 1 85 [54] 超声 PFOS 20 200 kHz,8 W·cm−2 1 60 [54] 微波+超声 PFOA 10 19.5 kHz,90 W 0.025 59 [56] 光催化+超声 PFOA 120 40 kHz, 500 W 7 65—70 [59] S2O82−+超声 NFDOHA 50 28 kHz,200 W 24 93.5 [57] S2O82−+超声 AFPO 46.4 20 kHz,300 W 2 51.2 [60] HCO3−+超声 PFOA 120 40 kHz,150 W 2 100 [58]
下载: 导出CSV
表 5 不同光催化剂对PFOA的降解效果
Table 5. PFOA degradation by using different photocatalysts
催化剂
Catalysts光源
Light source催化剂用量/
(mg·L−1)
Catalyst
dosage污染物起始浓度/
(mg·L−1)
Initial pollutant
concent-ration反应时间/h
Reac-tion time降解效率/%
Degra-dation efficiency脱氟率/%
Deflu-orination rate参考文献
References波长/nm
Wave length功率/W
Power类型
TypeNH4NO3-In2O3 NR. 350 Xe灯 125 0.41 6 57.8 36.1 [68] Fe3+/In2O3-600 254 32 NR. 2000 10 8 100 60 [69] In2O3纳米颗粒 254 15 汞灯 500 30 1.5 89.47 97.91 [70] In-Ga2O3 NR. 200 汞灯 500 20 1 100 NR. [72] Sheaf-like Ga2O3 254 14 汞灯 500 0.5 3 100 61 [73] BiOCl纳米片 365 500 汞灯 500 49.7 4 100 41.0 [74] BiOI0.95Br0.05 NR. 300 汞灯 400 20 2 96 NR. [75] InOOH 254 18 UV灯 250 20 3 83.4 NR. [67] BN 254 4 NR. 2500 50 4 100 52 [76] NR.,未报道. NR. , not reported.
下载: 导出CSV
表 6 不同方法产生水合电子对PFCAs的去除效果
Table 6. The removal effect of PFCAs by different methods of generation hydrated electron
体系
Methods污染物
Contaminate起始浓度/(μmol·L−1)
Initial concentration紫外光波长和功率/(nm&W)
UV light wavelength
and power反应时间/h
Reaction time降解效率/%
Degradation
rate脱氟率/%
Defluorination
rate参考文献
ReferencesUV/S PFCAs 25 NR.&18 48 100 ~55 [85] UV/S PFOA 20 254&10 24 100 88.5 [86] UV/S PFOA 4.83 200-400&450 0.5 NR. 82 [87] UV/I PFOA 25 NR.&15 6 93.9 76.8 [79] UV/I PFOA 30 254&14 10 99.8 72 [88] UV/S+I PFCAs 25 254&18 24 100 73—100 [84] NR.,未报道. NR. , not reported.
下载: 导出CSV
表 7 不同方法产生水合电子对PFSAs的去除效果
Table 7. The removal effect of PFSAs by different methods of generation hydrated electron
体系
Methods污染物
Contaminate起始浓度/(μmol·L−1)
Initial concentration紫外光波长和功率/(nm&W)
UV light wavelength and power反应时间/h
Reaction time降解效率/%
Degradation
rate脱氟率/%
Defluorination
rate参考文献
ReferencesUV/S PFOS 37.2 254&10 4 85.8 64.6 [80] UV/S PFOS 32 200—600&250 0.5 98 57 [90] UV/I PFOS 0.24 254&8 2.5 98 NR. [91] UV/I PFOS 30 254&14 10 73.9 44.4 [92] UV/NTA PFOS 25 254&14 10 85.4 46.8 [93] UV/EDTA PFOS 25 254&14 10 78.1 51.2 [94] NR.,未报道. NR. , not reported.
下载: 导出CSV
表 8 不同去除技术的优缺点对比
Table 8. Comparison of advantages and disadvantages of different removal methods
去除方法
Removal methods优点
Advantages缺点
Disadvantages高级氧化法 降解效率高、反应速率快 处理过程产生短链PFAS对环境有危害,短时间无法彻底矿化,反应能量
消耗多高级还原法 降解效率高、脱氟率高 水合电子pH受限,使用范围窄,需要加入额外化学试剂,无法大规模应用. 微生物降解法 绿色环保、可以处理不饱和PFAS 降解周期长,矿化程度有限,降解效率受污染物氟化程度影响.
下载: 导出CSV
-
[1] BUCK R C, FRANKLIN J, BERGER U, et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins[J]. Integrated Environmental Assessment and Management, 2011, 7(4): 513-541. doi: 10.1002/ieam.258 [2] GLÜGE J, SCHERINGER M, COUSINS I T, et al. An overview of the uses of per- and polyfluoroalkyl substances (PFAS)[J]. Environmental Science. Processes & Impacts, 2020, 22(12): 2345-2373. [3] GALLEN C, DRAGE D, EAGLESHAM G, et al. Australia-wide assessment of perfluoroalkyl substances (PFASs) in landfill leachates[J]. Journal of Hazardous Materials, 2017, 331: 132-141. doi: 10.1016/j.jhazmat.2017.02.006 [4] GALLEN C, EAGLESHAM G, DRAGE D, et al. A mass estimate of perfluoroalkyl substance (PFAS) release from Australian wastewater treatment plants[J]. Chemosphere, 2018, 208: 975-983. doi: 10.1016/j.chemosphere.2018.06.024 [5] HOUTZ E F, HIGGINS C P, FIELD J A, et al. Persistence of perfluoroalkyl acid precursors in AFFF-impacted groundwater and soil[J]. Environmental Science & Technology, 2013, 47(15): 8187-8195. [6] SUN M, AREVALO E, STRYNAR M, et al. Legacy and emerging perfluoroalkyl substances are important drinking water contaminants in the cape fear river watershed of north Carolina[J]. Environmental Science & Technology Letters, 2016, 3(12): 415-419. [7] LANGBERG H A, ARP H P H, BREEDVELD G D, et al. Paper product production identified as the main source of per- and polyfluoroalkyl substances (PFAS) in a Norwegian Lake: Source and historic emission tracking[J]. Environmental Pollution, 2020, 273: 116259. [8] DAUCHY X, BOITEUX V, COLIN A, et al. Deep seepage of per- and polyfluoroalkyl substances through the soil of a firefighter training site and subsequent groundwater contamination[J]. Chemosphere, 2019, 214: 729-737. doi: 10.1016/j.chemosphere.2018.10.003 [9] 范雨晴, 王雄, 陈铭杰, 等. 全氟和多氟烷基物质在北部湾海域表层沉积物中的污染特征及风险评估[J]. 环境化学, 2023, 42(3): 873-883. doi: 10.7524/j.issn.0254-6108.2022070804 FAN Y Q, WANG X, CHEN M J, et al. Pollution characteristics and risk assessment of perfluoroalkyl substances in surface sediments of the Beibu Gulf[J]. Environmental Chemistry, 2023, 42(3): 873-883 (in Chinese). doi: 10.7524/j.issn.0254-6108.2022070804
[10] GEBBINK W A, BOSSI R, RIGÉT F F, et al. Observation of emerging per- and polyfluoroalkyl substances (PFASs) in Greenland marine mammals[J]. Chemosphere, 2016, 144: 2384-2391. doi: 10.1016/j.chemosphere.2015.10.116 [11] 刘逸飞, 李阳, 赵楠楠, 等. 北京市售动物源性食品中全氟化合物赋存及居民摄入风险评估[J]. 环境化学, 2021, 40(11): 3360-3367. doi: 10.7524/j.issn.0254-6108.2022070804 LIU Y F, LI Y, ZHAO N N, et al. Occurrence of perfluoroalkyl substances in animal-derived food in Beijing and risk assessment of residents' intake[J]. Environmental Chemistry, 2021, 40(11): 3360-3367 (in Chinese). doi: 10.7524/j.issn.0254-6108.2022070804
[12] 胡佳玥, 戴家银. 全氟及多氟类化合物在人体分布及其毒性研究进展[J]. 生态毒理学报, 2013, 8(5): 650-657. doi: 10.7524/AJE.1673-5897.20130423002 HU J Y, DAI J Y. Advance in studies on human distribution and toxic effects of perfluoroalkyl and polyfluoroalkyl substances[J]. Asian Journal of Ecotoxicology, 2013, 8(5): 650-657 (in Chinese) . doi: 10.7524/AJE.1673-5897.20130423002
[13] 罗聪聪, 林佩娟, 杨育妮, 等. 全氟及多氟类化合物与卵巢癌关系的研究进展[J]. 国际妇产科学杂志, 2019, 46(6): 674-678. LUO C C, LIN P J, YANG Y N, et al. Research progress of the relationship between perfluoroalkyl and polyfluoroalkyl substances and ovarian cancer[J]. Journal of International Obstetrics and Gynecology, 2019, 46(6): 674-678 (in Chinese) .
[14] FISHER M, ARBUCKLE T E, WADE M K, et al. Do perfluoroalkyl substances affect metabolic function and plasma lipids?—Analysis of the 2007–2009, Canadian Health Measures Survey (CHMS) Cycle 1[J]. Environmental Research, 2013, 121: 95-103. doi: 10.1016/j.envres.2012.11.006 [15] LOPEZ-ESPINOSA M J, MONDAL D, ARMSTRONG B, et al. Thyroid function and perfluoroalkyl acids in children living near a chemical plant[J]. Environmental Health Perspectives, 2012, 120(7): 1036-1041. doi: 10.1289/ehp.1104370 [16] GRANDJEAN P, ANDERSEN E W, BUDTZ-JØRGENSEN E, et al. Serum vaccine antibody concentrations in children exposed to perfluorinated compounds[J]. JAMA, 2012, 307(4): 391-397. [17] AWAD R, ZHOU Y H, NYBERG E, et al. Correction: Emerging per- and polyfluoroalkyl substances (PFAS) in human milk from Sweden and China[J]. Environmental Science:Processes & Impacts, 2021, 23(1): 188. [18] LAM N H, CHO C R, KANNAN K, et al. A nationwide survey of perfluorinated alkyl substances in waters, sediment and biota collected from aquatic environment in Vietnam: Distributions and bioconcentration profiles[J]. Journal of Hazardous Materials, 2017, 323: 116-127. doi: 10.1016/j.jhazmat.2016.04.010 [19] JIN Y H, LIU W, SATO I, et al. PFOS and PFOA in environmental and tap water in China[J]. Chemosphere, 2009, 77(5): 605-611. doi: 10.1016/j.chemosphere.2009.08.058 [20] YAMAZAKI E, FALANDYSZ J, TANIYASU S, et al. Perfluorinated carboxylic and sulphonic acids in surface water media from the regions of Tibetan Plateau: Indirect evidence on photochemical degradation?[J]. Journal of Environmental Science and Health, Part A, 2016, 51(1): 63-69. doi: 10.1080/10934529.2015.1079113 [21] GE H, YAMAZAKI E, TANIYASU S, et al. Perfluoro alkyl substances in atomospheric particulate matters-size specific distribution analysis[J]. Organohalogen Compounds, 2015, 77: 492-495. [22] WANG B, YAO Y M, WANG Y, et al. Per- and polyfluoroalkyl substances in outdoor and indoor dust from China's mainland: Contributions of unknown precursors and implications for human exposure[J]. Environmental Science & Technology, 2022, 56(10): 6036-6045. [23] 陈森, 王新皓, 徐翊宸, 等. 市政污水处理系统中不同工艺段多氟/全氟烷基化合物(PFASs)的赋存、转化和去除[J]. 环境化学, 2023, 42(7): 2228-2241. CHEN S, WANG X H, XU Y C, et al. , Review on the occurrence, transformation and removal of per- and polyfluoroalkyl substances (PFASs) in different process segments of sewage wastewater treatment systems[J]. Environmental Chemistry, 2023, 42(7): 2228-2241(in Chinese).
[24] CHEN J J, TANG L B, CHEN W Q, et al. Flows, stock, and emissions of poly- and perfluoroalkyl substances in California carpet in 2000–2030 under different scenarios[J]. Environmental Science & Technology, 2020, 54(11): 6908-6918. [25] HORI H, NAGAOKA Y, MURAYAMA M, et al. Efficient decomposition of perfluorocarboxylic acids and alternative fluorochemical surfactants in hot water[J]. Environmental Science & Technology, 2008, 42(19): 7438-7443. [26] LUTZE H V, BREKENFELD J, NAUMOV S, et al. Degradation of perfluorinated compounds by sulfate radicals–New mechanistic aspects and economical considerations[J]. Water Research, 2017, 129: 509-519. [27] YANG S W, CHENG J H, SUN J, et al. Defluorination of aqueous perfluorooctanesulfonate by activated persulfate oxidation[J]. PLoS One, 2013, 8(10): e74877. doi: 10.1371/journal.pone.0074877 [28] LEE Y C, LO S L, CHIUEH P T, et al. Efficient decomposition of perfluorocarboxylic acids in aqueous solution using microwave-induced persulfate[J]. Water Research, 2009, 43(11): 2811-2816. doi: 10.1016/j.watres.2009.03.052 [29] HUIE R E, CLIFTON C L. Rate constants for hydrogen abstraction reactions of the sulfate radical, SO4−. Alkanes and ethers[J]. International Journal of Chemical Kinetics, 1989, 21(8): 611-619. doi: 10.1002/kin.550210802 [30] BUXTON G V, BYDDER M, ARTHUR SALMON G. The reactivity of chlorine atoms in aqueous solution Part Ⅱ. The equilibrium SO4−+Cl−Cl−Nsbd+SO42−[J]. Physical Chemistry Chemical Physics, 1999, 1(2): 269-273. doi: 10.1039/a807808d [31] YU X Y, BAO Z C, BARKER J R. Free radical reactions involving Cl·, and Cl2−·, and SO4−· in the 248 nm photolysis of aqueous solutions containing S2O82− and Cl−[J]. ChemInform, 2004, 35(14): no. [32] LIU C S, HIGGINS C P, WANG F, et al. Effect of temperature on oxidative transformation of perfluorooctanoic acid (PFOA) by persulfate activation in water[J]. Separation and Purification Technology, 2012, 91: 46-51. doi: 10.1016/j.seppur.2011.09.047 [33] HOUSE D A. Kinetics and mechanism of oxidations by peroxydisulfate[J]. Chemical Reviews, 1962, 62(3): 185-203. doi: 10.1021/cr60217a001 [34] LIANG C J, BRUELL C J, MARLEY M C, et al. Persulfate oxidation for in situ remediation of TCE. II. Activated by chelated ferrous ion[J]. Chemosphere, 2004, 55(9): 1225-1233. doi: 10.1016/j.chemosphere.2004.01.030 [35] LEE Y C, LO S L, CHIUEH P T, et al. Microwave-hydrothermal decomposition of perfluorooctanoic acid in water by iron-activated persulfate oxidation[J]. Water Research, 2010, 44(3): 886-892. doi: 10.1016/j.watres.2009.09.055 [36] HORI H, NAGAOKA Y, YAMAMOTO A, et al. Efficient decomposition of environmentally persistent perfluorooctanesulfonate and related fluorochemicals using zerovalent iron in subcritical water[J]. Environmental Science & Technology, 2006, 40(3): 1049-1054. [37] CHAPLIN B P. Critical review of electrochemical advanced oxidation processes for water treatment applications[J]. Environmental Science:Processes & Impacts, 2014, 16(6): 1182-1203. [38] ARIAS ESPANA V A, MALLAVARAPU M, NAIDU R. Treatment technologies for aqueous perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA): A critical review with an emphasis on field testing[J]. Environmental Technology & Innovation, 2015, 4: 168-181. [39] TROJANOWICZ M, BOJANOWSKA-CZAJKA A, BARTOSIEWICZ I, et al. Advanced Oxidation/Reduction Processes treatment for aqueous perfluorooctanoate (PFOA) and perfluorooctanesulfonate (PFOS)–A review of recent advances[J]. Chemical Engineering Journal, 2018, 336: 170-199. doi: 10.1016/j.cej.2017.10.153 [40] CARTER K E, FARRELL J. Oxidative destruction of perfluorooctane sulfonate using boron-doped diamond film electrodes[J]. Environmental Science & Technology, 2008, 42(16): 6111-6115. [41] ZHUO Q F, DENG S B, YANG B, et al. Efficient electrochemical oxidation of perfluorooctanoate using a Ti/SnO2-Sb-Bi anode[J]. Environmental Science & Technology, 2011, 45(7): 2973-2979. [42] 滕影, 王雯冉, 黄柳青, 等. 全氟烷基化合物的去除技术研究进展[J]. 环境化学, 2023, 42(7): 2210-2227. TENG Y, WANG W R, HUANG L Q, et al. , Research progress on the removal of perfluorinated allkyl substances: A review[J]. Environmental Chemistry, 2023, 42(7): 2210-2227(in Chinese).
[43] ZHUO Q F, DENG S B, YANG B, et al. Degradation of perfluorinated compounds on a boron-doped diamond electrode[J]. Electrochimica Acta, 2012, 77: 17-22. doi: 10.1016/j.electacta.2012.04.145 [44] YANG B, WANG J B, JIANG C J, et al. Electrochemical mineralization of perfluorooctane sulfonate by novel F and Sb co-doped Ti/SnO2 electrode containing Sn-Sb interlayer[J]. Chemical Engineering Journal, 2017, 316: 296-304. doi: 10.1016/j.cej.2017.01.105 [45] NIU J F, LIN H, XU J L, et al. Electrochemical mineralization of perfluorocarboxylic acids (PFCAs) by Ce-doped modified porous nanocrystalline PbO2 film electrode[J]. Environmental Science & Technology, 2012, 46(18): 10191-10198. [46] HWANG J H, LI SIP Y Y, KIM K T, et al. Nanoparticle-embedded hydrogel synthesized electrodes for electrochemical oxidation of perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS)[J]. Chemosphere, 2022, 296: 134001. doi: 10.1016/j.chemosphere.2022.134001 [47] ZHANG C H, TANG J W, PENG C, et al. Degradation of perfluorinated compounds in wastewater treatment plant effluents by electrochemical oxidation with Nano-ZnO coated electrodes[J]. Journal of Molecular Liquids, 2016, 221: 1145-1150. doi: 10.1016/j.molliq.2016.06.093 [48] XIAO H S, LV B Y, ZHAO G H, et al. Hydrothermally enhanced electrochemical oxidation of high concentration refractory perfluorooctanoic acid[J]. The Journal of Physical Chemistry A, 2011, 115(47): 13836-13841. doi: 10.1021/jp207519j [49] SCHAEFER C E, ANDAYA C, BURANT A, et al. Electrochemical treatment of perfluorooctanoic acid and perfluorooctane sulfonate: Insights into mechanisms and application to groundwater treatment[J]. Chemical Engineering Journal, 2017, 317: 424-432. doi: 10.1016/j.cej.2017.02.107 [50] ZHUO Q F, LUO M Q, GUO Q W, et al. Electrochemical oxidation of environmentally persistent perfluorooctane sulfonate by a novel lead dioxide anode[J]. Electrochimica Acta, 2016, 213: 358-367. doi: 10.1016/j.electacta.2016.07.005 [51] ZHAO H Y, GAO J X, ZHAO G H, et al. Fabrication of novel SnO2-Sb/carbon aerogel electrode for ultrasonic electrochemical oxidation of perfluorooctanoate with high catalytic efficiency[J]. Applied Catalysis B:Environmental, 2013, 136/137: 278-286. doi: 10.1016/j.apcatb.2013.02.013 [52] VECITIS C D, PARK H, CHENG J, et al. Kinetics and mechanism of the sonolytic conversion of the aqueous perfluorinated surfactants, perfluorooctanoate (PFOA), and perfluorooctane sulfonate (PFOS) into inorganic products[J]. The Journal of Physical Chemistry A, 2008, 112(18): 4261-4270. doi: 10.1021/jp801081y [53] McNAMARA W B, DIDENKO Y T, SUSLICK K S. Pressure during sonoluminescence[J]. The Journal of Physical Chemistry B, 2003, 107(30): 7303-7306. doi: 10.1021/jp034236b [54] RODRIGUEZ-FREIRE L, ABAD-FERNÁNDEZ N, SIERRA-ALVAREZ R, et al. Sonochemical degradation of perfluorinated chemicals in aqueous film-forming foams[J]. Journal of Hazardous Materials, 2016, 317: 275-283. doi: 10.1016/j.jhazmat.2016.05.078 [55] CAMPBELL T, HOFFMANN M R. Sonochemical degradation of perfluorinated surfactants: Power and multiple frequency effects[J]. Separation and Purification Technology, 2015, 156: 1019-1027. doi: 10.1016/j.seppur.2015.09.053 [56] HORIKOSHI S, SATO S, ABE M, et al. A novel liquid plasma AOP device integrating microwaves and ultrasounds and its evaluation in defluorinating perfluorooctanoic acid in aqueous media[J]. Ultrasonics Sonochemistry, 2011, 18(5): 938-942. doi: 10.1016/j.ultsonch.2011.01.003 [57] HORI H, NAGANO Y, MURAYAMA M, et al. Efficient decomposition of perfluoroether carboxylic acids in water with a combination of persulfate oxidant and ultrasonic irradiation[J]. Journal of Fluorine Chemistry, 2012, 141: 5-10. doi: 10.1016/j.jfluchem.2012.05.012 [58] PHAN THI L A, DO H T, LO S L. Enhancing decomposition rate of perfluorooctanoic acid by carbonate radical assisted sonochemical treatment[J]. Ultrasonics Sonochemistry, 2014, 21(5): 1875-1880. doi: 10.1016/j.ultsonch.2014.03.027 [59] PANCHANGAM S C, LIN A Y C, TSAI J H, et al. Sonication-assisted photocatalytic decomposition of perfluorooctanoic acid[J]. Chemosphere, 2009, 75(5): 654-660. doi: 10.1016/j.chemosphere.2008.12.065 [60] HAO F F, GUO W L, WANG A Q, et al. Intensification of sonochemical degradation of ammonium perfluorooctanoate by persulfate oxidant[J]. Ultrasonics Sonochemistry, 2014, 21(2): 554-558. doi: 10.1016/j.ultsonch.2013.09.016 [61] MORIWAKI H, TAKAGI Y, TANAKA M, et al. Sonochemical decomposition of perfluorooctane sulfonate and perfluorooctanoic acid[J]. Environmental Science & Technology, 2005, 39(9): 3388-3392. [62] YOU X, YU L L, XIAO F F, et al. Synthesis of phosphotungstic acid-supported bimodal mesoporous silica-based catalyst for defluorination of aqueous perfluorooctanoic acid under vacuum UV irradiation[J]. Chemical Engineering Journal, 2018, 335: 812-821. doi: 10.1016/j.cej.2017.10.123 [63] DO H T, PHAN THI L A, DAO NGUYEN N H, et al. Tailoring photocatalysts and elucidating mechanisms of photocatalytic degradation of perfluorocarboxylic acids (PFCAs) in water: A comparative overview[J]. Journal of Chemical Technology & Biotechnology, 2020, 95(10): 2569-2578. [64] WANG Y, ZHANG P Y. Photocatalytic decomposition of perfluorooctanoic acid (PFOA) by TiO2 in the presence of oxalic acid[J]. Journal of Hazardous Materials, 2011, 192(3): 1869-1875. doi: 10.1016/j.jhazmat.2011.07.026 [65] CHEN M J, LO S L, LEE Y C, et al. Photocatalytic decomposition of perfluorooctanoic acid by transition-metal modified titanium dioxide[J]. Journal of Hazardous Materials, 2015, 288: 168-175. doi: 10.1016/j.jhazmat.2015.02.004 [66] HORI H, ISHIDA K, INOUE N, et al. Photocatalytic mineralization of hydroperfluorocarboxylic acids with heteropolyacid H4SiW12O40 in water[J]. Chemosphere, 2011, 82(8): 1129-1134. doi: 10.1016/j.chemosphere.2010.11.038 [67] XU J J, WU M M, YANG J W, et al. Efficient photocatalytic degradation of perfluorooctanoic acid by a wide band gap p-block metal oxyhydroxide InOOH[J]. Applied Surface Science, 2017, 416: 587-592. doi: 10.1016/j.apsusc.2017.04.040 [68] LIU J Q, GUO C J, WU N N, et al. Efficient photocatalytic degradation of PFOA in N-doped In2O3/simulated sunlight irradiation system and its mechanism[J]. Chemical Engineering Journal, 2022, 435: 134627. doi: 10.1016/j.cej.2022.134627 [69] LIU X Q, CHEN Z J, TIAN K, et al. Fe3+ promoted the photocatalytic defluorination of perfluorooctanoic acid (PFOA) over In2O3[J]. ACS ES& T Water, 2021, 1(11): 2431-2439. [70] ZHANG W L, EFSTATHIADIS H, LI L Y, et al. Environmental factors affecting degradation of perfluorooctanoic acid (PFOA) by In2O3 nanoparticles[J]. Journal of Environmental Sciences, 2020, 93: 48-56. doi: 10.1016/j.jes.2020.02.028 [71] WU Y Y, LI Y Q, FANG C, et al. Highly efficient degradation of perfluorooctanoic acid over a MnOx-modified oxygen-vacancy-rich In2O3 photocatalyst[J]. ChemCatChem, 2019, 11(9): 2297-2303. doi: 10.1002/cctc.201900273 [72] TAN X J, CHEN G H, XING D Y, et al. Indium-modified Ga2O3 hierarchical nanosheets as efficient photocatalysts for the degradation of perfluorooctanoic acid[J]. Environmental Science:Nano, 2020, 7(8): 2229-2239. doi: 10.1039/D0EN00259C [73] SHAO T, ZHANG P Y, JIN L, et al. Photocatalytic decomposition of perfluorooctanoic acid in pure water and sewage water by nanostructured gallium oxide[J]. Applied Catalysis B:Environmental, 2013, 142/143: 654-661. doi: 10.1016/j.apcatb.2013.05.074 [74] WU Y Y, HU Y X, HAN M Q, et al. Mechanism insights into the facet-dependent photocatalytic degradation of perfluorooctanoic acid on BiOCl nanosheets[J]. Chemical Engineering Journal, 2021, 425: 130672. doi: 10.1016/j.cej.2021.130672 [75] LI T F, WANG C S, WANG T C, et al. Highly efficient photocatalytic degradation toward perfluorooctanoic acid by bromine doped BiOI with high exposure of (001) facet[J]. Applied Catalysis B:Environmental, 2020, 268: 118442. doi: 10.1016/j.apcatb.2019.118442 [76] DUAN L J, WANG B, HECK K, et al. Efficient photocatalytic PFOA degradation over boron nitride[J]. Environmental Science & Technology Letters, 2020, 7(8): 613-619. [77] REN Z F, BERGMANN U, LEIVISKÄ T. Reductive degradation of perfluorooctanoic acid in complex water matrices by using the UV/sulfite process[J]. Water Research, 2021, 205: 117676. doi: 10.1016/j.watres.2021.117676 [78] CUI J K, GAO P P, DENG Y. Destruction of per- and polyfluoroalkyl substances (PFAS) with advanced reduction processes (ARPs): A critical review[J]. Environmental Science & Technology, 2020, 54(7): 3752-3766. [79] QU Y, ZHANG C J, LI F, et al. Photo-reductive defluorination of perfluorooctanoic acid in water[J]. Water Research, 2010, 44(9): 2939-2947. doi: 10.1016/j.watres.2010.02.019 [80] GU Y R, LIU T Z, WANG H J, et al. Hydrated electron based decomposition of perfluorooctane sulfonate (PFOS) in the VUV/sulfite system[J]. Science of the Total Environment, 2017, 607/608: 541-548. doi: 10.1016/j.scitotenv.2017.06.197 [81] GU Y R, DONG Z J, CHEN H J, et al. Efficient decomposition of perfluorinated compounds in a high photon flux UV/sulfite system[J]. IOP Conference Series:Earth and Environmental Science, 2020, 546(4): 042027. doi: 10.1088/1755-1315/546/4/042027 [82] GU Y R, LIU T Z, ZHANG Q, et al. Efficient decomposition of perfluorooctanoic acid by a high photon flux UV/sulfite process: Kinetics and associated toxicity[J]. Chemical Engineering Journal, 2017, 326: 1125-1133. doi: 10.1016/j.cej.2017.05.156 [83] CAO M H, WANG B B, YU H S, et al. Photochemical decomposition of perfluorooctanoic acid in aqueous periodate with VUV and UV light irradiation[J]. Journal of Hazardous Materials, 2010, 179(1/2/3): 1143-1146. [84] LIU Z K, CHEN Z H, GAO J Y, et al. Accelerated degradation of perfluorosulfonates and perfluorocarboxylates by UV/sulfite + iodide: Reaction mechanisms and system efficiencies[J]. Environmental Science & Technology, 2022, 56(6): 3699-3709. [85] BENTEL M J, YU Y C, XU L H, et al. Defluorination of per- and polyfluoroalkyl substances (PFASs) with hydrated electrons: Structural dependence and implications to PFAS remediation and management[J]. Environmental Science & Technology, 2019, 53(7): 3718-3728. [86] SONG Z, TANG H Q, WANG N, et al. Reductive defluorination of perfluorooctanoic acid by hydrated electrons in a sulfite-mediated UV photochemical system[J]. Journal of Hazardous Materials, 2013, 262: 332-338. doi: 10.1016/j.jhazmat.2013.08.059 [87] ABUSALLOUT I, WANG J L, HANIGAN D. Emerging investigator series: Rapid defluorination of 22 per- and polyfluoroalkyl substances in water using sulfite irradiated by medium-pressure UV[J]. Environmental Science:Water Research & Technology, 2021, 7(9): 1552-1562. [88] GUO C X, ZHANG C J, SUN Z Y, et al. Synergistic impact of humic acid on the photo-reductive decomposition of perfluorooctanoic acid[J]. Chemical Engineering Journal, 2019, 360: 1101-1110. doi: 10.1016/j.cej.2018.10.204 [89] BENTEL M J, YU Y C, XU L H, et al. Degradation of perfluoroalkyl ether carboxylic acids with hydrated electrons: Structure–reactivity relationships and environmental implications[J]. Environmental Science & Technology, 2020, 54(4): 2489-2499. [90] GU Y R, DONG W Y, LUO C, et al. Efficient reductive decomposition of perfluorooctanesulfonate in a high photon flux UV/sulfite system[J]. Environmental Science & Technology, 2016, 50(19): 10554-10561. [91] PARK H, VECITIS C D, CHENG J, et al. Reductive defluorination of aqueous perfluorinated alkyl surfactants: Effects of ionic headgroup and chain length[J]. The Journal of Physical Chemistry A, 2009, 113(4): 690-696. doi: 10.1021/jp807116q [92] SUN Z Y, ZHANG C J, CHEN P, et al. Impact of humic acid on the photoreductive degradation of perfluorooctane sulfonate (PFOS) by UV/Iodide process[J]. Water Research, 2017, 127: 50-58. doi: 10.1016/j.watres.2017.10.010 [93] SUN Z Y, ZHANG C J, XING L, et al. UV/nitrilotriacetic acid process as a novel strategy for efficient photoreductive degradation of perfluorooctanesulfonate[J]. Environmental Science & Technology, 2018, 52(5): 2953-2962. [94] GU P F, ZHANG C J, SUN Z Y, et al. Enhanced photoreductive degradation of perfluorooctanesulfonate by UV irradiation in the presence of ethylenediaminetetraacetic acid[J]. Chemical Engineering Journal, 2020, 379: 122338. doi: 10.1016/j.cej.2019.122338 [95] HUANG S, JAFFÉ P R. Defluorination of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) by Acidimicrobium sp. strain A6[J]. Environmental Science & Technology, 2019, 53(19): 11410-11419. [96] YU Y C, ZHANG K Y, LI Z, et al. Microbial cleavage of C–F bonds in two C6 per- and polyfluorinated compounds via reductive defluorination[J]. Environmental Science & Technology, 2020, 54(22): 14393-14402. [97] LIOU J S C, SZOSTEK B, DeRITO C M, et al. Investigating the biodegradability of perfluorooctanoic acid[J]. Chemosphere, 2010, 80(2): 176-183. doi: 10.1016/j.chemosphere.2010.03.009 [98] LUO Q, LU J H, ZHANG H, et al. Laccase-catalyzed degradation of perfluorooctanoic acid[J]. Environmental Science & Technology Letters, 2015, 2(7): 198-203. [99] DING R, WU Y, YANG F, et al. Degradation of low-concentration perfluorooctanoic acid via a microbial-based synergistic method: Assessment of the feasibility and functional microorganisms[J]. Journal of Hazardous Materials, 2021, 416: 125857. doi: 10.1016/j.jhazmat.2021.125857 [100] YU Y C, CHE S, REN C X, et al. Microbial defluorination of unsaturated per- and polyfluorinated carboxylic acids under anaerobic and aerobic conditions: A structure specificity study[J]. Environmental Science & Technology, 2022, 56(8): 4894-4904. [101] ZHAO J Y, ZHANG H, ZHAN J X, et al. Contrastive study on organic contaminated soils remediated using dielectric barrier discharge (DBD) plasma[J]. Separation and Purification Technology, 2023, 306: 122576. doi: 10.1016/j.seppur.2022.122576 [102] TRANG B, LI Y, XUE X S, et al. Low-temperature mineralization of perfluorocarboxylic acids[J]. Science, 2022, 377(6608): 839-845. doi: 10.1126/science.abm8868 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 9063
- HTML全文浏览数: 9063
- PDF下载数: 420
- 施引文献: 0


