

-
燃煤电站烟气排放到大气中的氮氧化物(NOx,包括NO、NO2在内),已被证明不仅会造成诸如雾霾、光化学污染和酸雨等一系列环境问题,还会对人的身体健康产生极大的危害[1]. 利用NH3还原NOx的选择性催化还原法(NH3-SCR)是目前最主流的NOx处理方式,除此还有储存还原法 (NSR)和选择性催化氧化吸收法(SCO). 对于传统的SCR脱硝路线,有研究表明烟气中存在的NO2能通过一种快速SCR机制(4NH3 + 2NO + 2NO2 → 4N2 + 6H2O)促进NO和NH3反应,且其反应速率比标准SCR反应(4NH3 + 4NO + O2 → 4N2 + 6H2O)快10倍[2]. 而氧化脱硝路线的关键就是将低溶解度的NO氧化为高溶解度的NO2或N2O5,再结合吸收法脱除NOx. 其中,本课题组提出的臭氧氧化多种污染物协同脱除技术可利用O3分子的强氧化性实现NO、Hg0、VOCs的一体化氧化脱除. 虽然已成功应用于多个超低排放改造工程[3],但不仅需要增加相应的O3发生设备,在实际使用时还要投入过量的O3以保证脱除效果,极大地增加了相应的运行成本. 而如果能利用烟气中残留的O2实现NO向NO2的转化,则理论上可节省2/3的O3消耗. 可见,NO向NO2的催化氧化在多种脱硝路线中起着关键作用,因此开发具有良好催化活性的NO催化氧化催化剂已成为一大研究热点.
过渡金属氧化物作为最常见的催化氧化材料,在很多领域已表现出优异的催化性能. 而以过渡金属为基底组成的双元或多元复合氧化物往往由于多种金属之间的相互作用而使催化剂具备优于单一金属氧化物的催化性能,目前受到广泛的研究关注[4]. 王先涛等[5]发现溶胶凝胶法制备的Mn-Ce-Ox催化剂中锰氧化物将以无定形的型态均匀地分散在催化剂表面,使催化剂具有更大的比表面积和良好的催化活性. Gao[6]通过XPS和DRIFTS发现由于Mn和Co间存在协同效应(Co2+ + Mn4+ ↔ Mn3+ + Co3+; Co3+ + Mn2+ ↔ Mn3+ + Co2+),制备的具有丰富高价态Mn和Co的Mn0.52Co2.48O4催化剂在250 ℃的NO氧化转化率可达90%(GHSV=
30000 h−1). Cao等[7]则采用等体积浸渍法制备了Fe-Mn/TiO2催化剂,发现适当的负载量和Mn/Fe能使活性组分更均匀地分布在载体孔道内,提升催化活性. Li等[8]也发现对其通过共沉淀法制备的无定形Mn-Co-Ce-Ox催化剂,适量配比的Ce掺杂才能达到最佳催化活性,随Ce掺杂量的增加NO 转化率先增加(0—20% wt)后降低(>30% wt). 除此Li等[9]进一步考察了反应温度、NO初始浓度和O2浓度对Mn-Co-Ce(20)-Ox催化剂活性的影响.尽管上述无定形的金属复合氧化物都具有较好的催化活性,但是它们在复杂烟气组分下活性往往受H2O和SO2影响较大. 对于NO的氧化转化,催化剂表面生成的硝酸盐物种是关键中间产物,H2O的存在会促进这些中间产物向更加稳定的硝酸盐转化,占据大量的活性位点造成失活. 而SO2在活性位点上结合生成的硫酸/亚硫酸盐则更加难以分解,将造成比H2O严重多倍的失活. 例如,Li等[9]制备的无定形Mn-Co-Ce-Ox催化剂在分别引入H2O和SO2后,催化活性在60 min后均出现严重下降. Jiang等[10]通过研究发现,SO2能通过双齿配位形式与Fe-Mn/TiO2催化剂上的活性位点结合生成单核硫酸盐,阻碍NO的吸附,造成NO转化率的严重下降. 而Lin[11]发现酸蚀处理后的LaMnO3+δ 催化剂的SO2耐受性得到了提高. 除此之外,采用不同合成方法、选取最佳煅烧温度及优化催化剂各组分配比都或许能在降低部分催化活性的前提下提升催化剂对复杂烟气环境的适应性. 本文采用一种柠檬酸溶胶凝胶法制备了系列锰钴铈复合金属氧化物,测试了它们用于催化O2氧化NO的活性,并利用多种表征手段对几种催化剂的理化性质进行了表征,探究了Mn/Co比对催化剂催化氧化NO活性的影响,以期为NO催化氧化催化剂的开发提供新的参考.
-
柠檬酸-溶胶凝胶法制备系列具备不同Mn/Co物质的量比的锰钴铈复合金属氧化物(MnCoCeOx)催化剂. 室温下取一定量的硝酸锰溶液(50% wt Mn(NO3)2)、硝酸钴(Co(NO3)2·6H2O)以及
2.6053 g硝酸亚铈(0.006 mmol Ce(NO3)2·6H2O) 、2.5 g 聚乙二醇(PEG 400)与30 mL去离子水搅拌混合,制得前驱体盐A溶液(其中Mn+Co+Ce=0.03 mmol). 称取12.6084 g柠檬酸(0.06 mmol C6H8O7·H2O)溶于60 mL去离子水,超声搅拌均匀后得柠檬酸盐B溶液. 将A、B溶液混合均匀在80 ℃、400 r·min−1磁力搅拌下蒸发至淡黄色透明胶体溶液生成,继续蒸发至沸腾有气泡后转移至110 ℃烘箱中烘干过夜. 所得固体置于500 ℃管式炉中煅烧4 h后制得不同金属物质的量比的MnCoCeOx催化剂. 根据不同Mn/Co物质的量比将4组催化剂分别命名为Mn3Co1Ce1、Mn2.5Co1.5Ce1、Mn2Co2Ce1、Mn1.5Co2.5Ce1. -
X射线多晶衍射仪(RIGAKU D/MAX
2550 /PC)辐射源为单色Cu Kα靶衍射(λ=0.15406 nm),最大输出功率为18 kW,测量区间为10°—80°. N2吸脱附曲线数据由全自动比表面积及孔隙度分析仪(Micromeritics ASAP2460 )测得,通过Brunauer-Emmett-Teller (BET)和Barrett-Joyner-Halenda (BJH)方法分别获得样品的比表面积、孔容和平均孔径. 光电子能谱仪(Thermo Scientific ESCALAB 250 Xi)搭配标准Al Kα辐射源(1486.6 eV),样品所有电子结合能根据标准C 1s峰(284.6 eV)进行校正. 氨气程序性升温脱附测试(NH3-TPD)在全自动程序升温化学吸附仪(Micromeritics AutoChem Ⅱ2920 )上进行,取0.05 g催化剂置于U形样品管中,300 ℃下用50 mL·min−1 He预处理1 h. 然后降温到50 ℃,在40 mL·min−1 NH3/He下吸附1 h,吸附后在50 mL·min−1He下吹扫1 h,直至NH3基线信号稳定后,以恒定的速率(10 ℃·min−1)升温到825 ℃,脱附信号由TCD检测器测得. 氢气程序升温还原测试(H2-TPR)也在同一台化学吸附仪上完成,取0.05 g催化剂置于U形样品管中,在300 ℃下条件下,用50 mL·min−1 He预处理1 h. 然后降温到50 ℃,切换到30 mL·min−1 10 % H2/Ar气氛,20 min后至H2基线信号稳定,以恒定的速率(10 ℃·min−1)升温到825 ℃,脱附信号由TCD检测器测得. -
催化剂催化O2氧化NO活性测试在固定床反应系统中进行. 试验时,称取0.1 g催化剂与一定量石英砂(40—60目)混合,以确保催化床层高度不变,随后填入有石英棉床层的不锈钢反应管中(内径8 mm),通过内嵌式K型热电偶实时监测反应温度. 反应气由钢瓶气(N2: 99.999%, O2: 99.999%,NO/N2: 0.2%,SO2/N2: 0.1%)供应,通过质量流量计组(Alicat Scientific, Inc.) 配置混气. NO的初始浓度为368 mg·m−3,O2浓度为10 %体积分数,N2作平衡气,总流量300 mL·min−1,体积空速(GHSV)约为
28000 h−1. 试验中烟气各组分的浓度(NO、NO2、N2O 和 H2O 等)由傅里叶红外在线烟气分析仪(Gasmet DX4000)实时测得,催化剂的NO氧化效率由式(1)计算得出:式中,η为NO的转化率,C[NO]in 、C[NO]out为反应管进出口NO的浓度,单位为mg·m−3.
-
具有不同Mn/Co物质的量比的4组MnCoCeOx催化剂催化O2氧化NO的催化活性结果如图1所示. 在测试温度(120—360 ℃)范围内,所有催化剂的NO转化效率均随着温度升高先增大,在260—280 ℃内达到最高点,之后开始下降,呈现出明显的火山状效率曲线. 且各催化剂在高温段(280—360 ℃)内转化效率均无明显差别,并在280 ℃时都拥有79%以上NO转化率,说明其反应活性在高温段主要受如图1中平衡曲线所示2NO + O2 = 2NO2的热力学限制 [12]. 在低温段(120—280 ℃)各催化剂的NO转化效率则展现出明显差异. 其中,Mn2.5Co1.5Ce1催化剂具备最佳活性,在260 ℃达到了最高的NO转化率83.9%;同时在250—300 ℃温度区间内NO转化率均大于80%,温度适应性良好. 除此,其达到最大转化率的温度(260 ℃)相较于其他3种催化剂(280 ℃)也更低. 本文制备的Mn2.5Co1.5Ce1催化剂与文献报道的几种锰基催化剂催化氧化NO的性能对比如表1所示. 从表1 可看出,本文制备的MnCoCeOx催化剂具有良好的催化性能. 表1中各催化剂间的对比也表明了掺杂Co能有效提升在锰基催化剂催化性能. 总的来说,4组不同物质的量比的MnCoCeOx催化剂催化活性大小顺序为Mn2.5Co1.5Ce1> Mn3Co1Ce1> Mn1.5Co2.5Ce1> Mn2Co2Ce1. 可以看到,随着Mn/Co比的减小,MnCoCeOx催化剂催化活性先升高后下降,说明适当的Mn/Co有利于提高MnCoCeOx催化剂催化氧化NO的活性. 但是值得注意的是,Mn1.5Co2.5Ce1催化剂的催化活性却比Mn2Co2Ce1催化剂更高,需要通过进一步表征探究Mn/Co比对MnCoCeOx催化剂催化活性的影响.
-
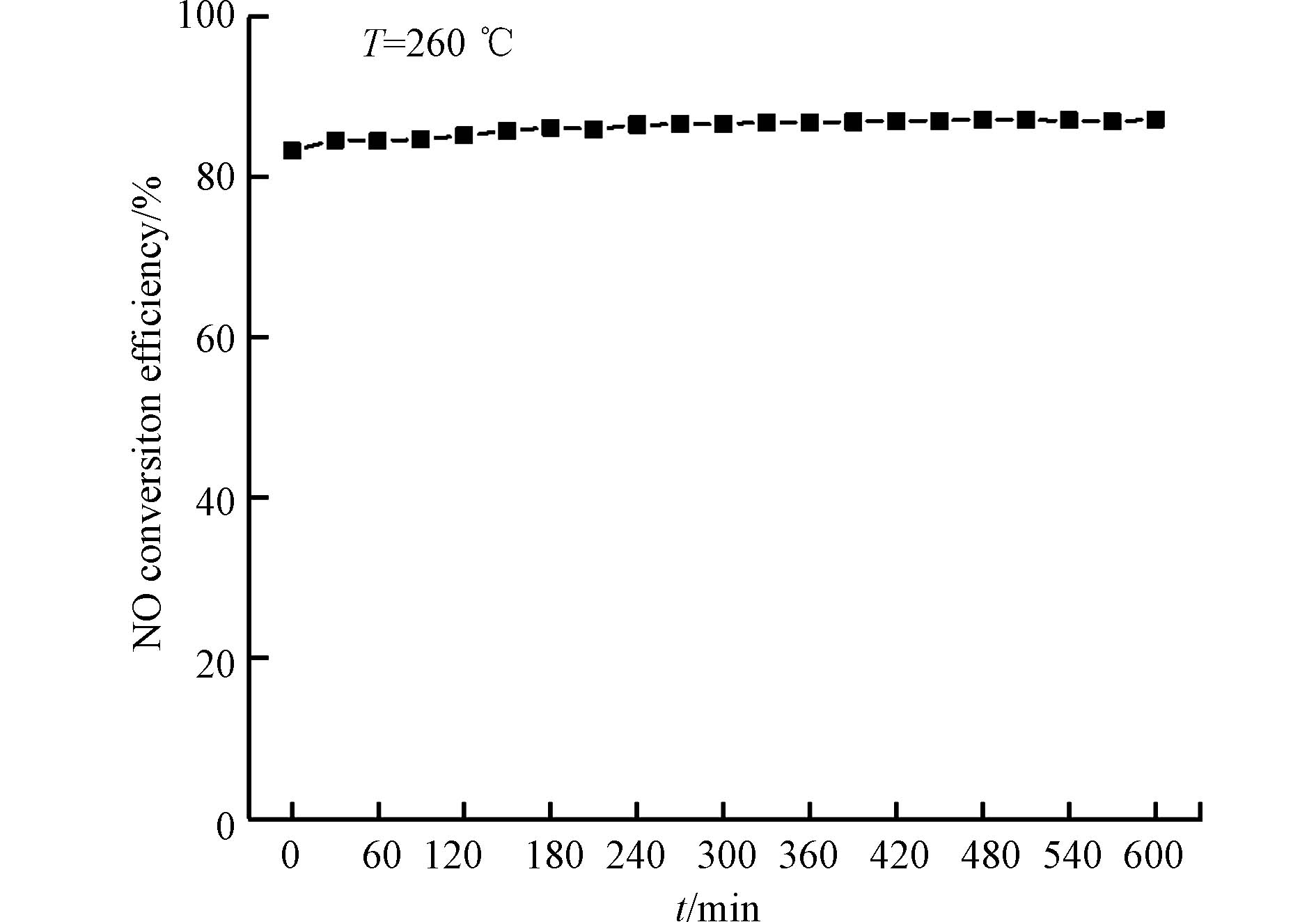
为进一步评估制备的锰钴铈复合氧化物催化剂的性能,选取催化活性最高的Mn2.5Co1.5Ce1催化剂进行了稳定性测试,试验条件与前文活性测试一致,试验结果如图2所示. 可以看到,在反应温度为260 ℃时,在600 min的反应时间内,Mn2.5Co1.5Ce1催化剂的NO转化率维持在84%—86%之间,表明Mn2.5Co1.5Ce1催化剂在具有良好的催化氧化NO性能的同时还具备了较高的稳定性.
-
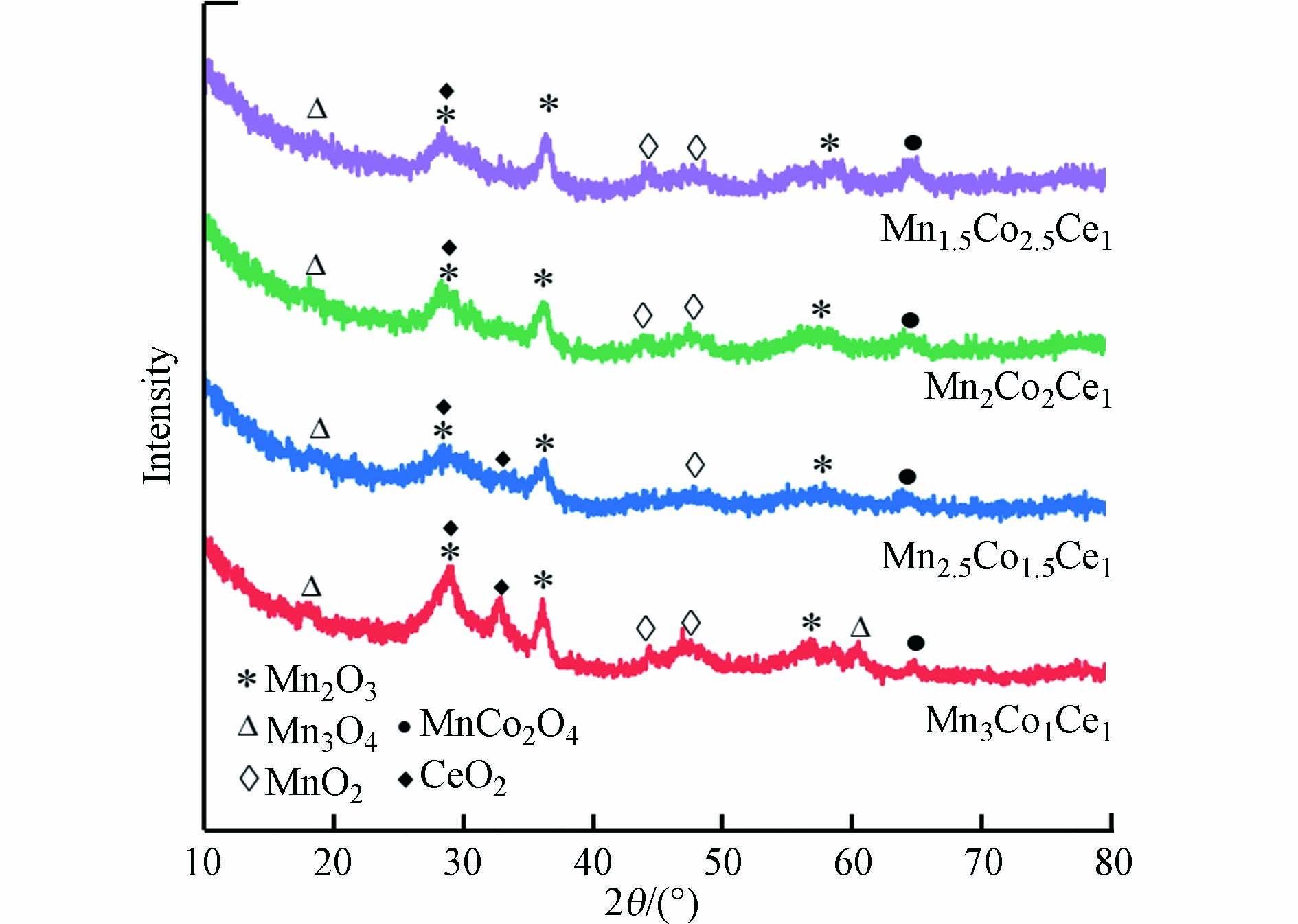
4组MnCoCeOx样品的XRD图谱如图3所示. 所有样品均没有尖锐的强衍射峰,呈现无定形,低结晶结构. 其中,强度相对较强的在(2θ)28.5°、33.1°和35.7°的特征衍射峰分别对应于和CeO2(PDF#43-
1002 )和Mn2O3(PDF#33-0900). 说明在制备得到的MnCoCeOx样品中,Ce和Mn分别主要以CeO2和Mn2O3的形态存在. 而对应这两种晶相的衍射峰都十分宽大,并随着Mn/Co的减小进一步变宽变弱,说明Co的增加有利于加强Mn-Ce 之间的相互作用[13]. 另外,4组催化剂样品的XRD图谱还出现对应于Mn3O4(PDF#18-0803)、MnO2(PDF#50-0866)的弱峰,表明样品中存在多种价态的Mn. 除此,在64.2°还出现对应于MnCo2O4(PDF#01-1130 )的弱峰,与文献对应一致[14],说明少量的Mn与Co形成了MnCo2O4的尖晶石颗粒. 有研究表明,在MnCo2O4尖晶石结构中Mn-Co之间存在协同作用,可暴露更多Co3+作为活性位点,使其具备良好的催化氧化能力[15],这可能是其具备更好催化活性的原因之一. -
为了探究4组MnCoCeOx样品的孔隙结构,应用了表征材料孔隙结构最有力的N2吸脱附测试手段,得到了如图4所示的N2吸脱附曲线和孔径分布结果. 发现所有样品的N2吸脱附曲线都属于带有H3型回滞环的第Ⅳ类等温线,表明其具有明显的介孔结构[16]. 孔径分布图则显示所有样品在约3.0 nm和约4.5 nm处有两个分布峰,表明催化剂富有的孔径分布集中均匀的小介孔.
表2给出了所有MnCoCeOx催化剂比表面积、孔容及平均孔径的具体数据. 可以看出,拥有最佳催化活性的Mn2.5Co1.5Ce1催化剂具有最大的比表面积(108.37 m2·g−1)、第二大的孔容(0.096 cm3·g−1)以及最小的平均孔径(5.50 nm). 一般来说,更大的比表面积可以提供更多的活性位点,较大的孔容则提供了优异的吸附性能,而更小的孔径则让吸附的反应物分子间更容易接触. 另外通过对比发现,不同Mn/Co比MnCoCeOx催化剂的比表面积、孔容和平均孔径大小排序都与它们的活性顺序相对应. 这说明较大的比表面积、孔容和较小的孔径可能是Mn2.5Co1.5Ce1催化剂具备良好催化活性的原因之一.
-
为了探究各催化剂的表面各元素价态的差异,进行XPS测试,结果如图5所示. 图5(a)为不同MnCoCeOx催化剂的Ce 3d图谱,其中Ce 3d曲线可以解卷积分为8个峰,分别为V(~882 eV)、V′(~884 eV)、V″(~888.4 eV)、V‴(~898.1 eV)、U(~900.7 eV)、U′(~902.1 eV)、U″(~906.7 eV)、U‴(~916.5 eV),所有Vx代表Ce 3d5/2自旋轨道分量,而所有Ux则代表Ce 3d3/2. 其中,V′和U′对应Ce3+物种,而其余所有峰V、V″、V‴、U、U″、U‴对应Ce4+物种[14,17 − 18]. 图5(b)展示了不同MnCoCeOx催化剂的Mn 2p图谱,采用同样的方法后可将Mn 2p3/2分为3个位于~641.1 eV,~642.3 eV,~644.0 eV的峰,分别对应于Mn2+、Mn3+、Mn4+物种[19 − 20]. 不同MnCoCeOx催化剂的Co 2p图谱如图5(c),图中Co 2p3/2 可按上述方法分为2个分别对应Co3+(~780.3 eV)、Co2+(781.6 eV)的主峰和一个对应于Co3O4(~786.4 eV)的强卫星峰[19,21 − 22]. 图5(d)是不同MnCoCeOx催化剂的O 1s图谱,解卷积后得到两个明显的分峰,分别对应于化学吸附氧Oad(531.2—532.2 eV)和晶格氧Ola(529.2—530.0 eV)[23 − 24].
表3是对各组催化剂样品的XPS图谱进行积分处理后得到的数据. 从表3可以看出,各催化剂中Ce4+和Mn3+是相应元素存在的主要形式,这与前面XRD分析一致. 通过对比发现Mn2.5Co1.5Ce1催化剂拥有最高比例的Mn3++Mn4+(87.52 %)和Co3+(64.35 %),而有研究表明高价态的Mn和Co都是有利于NO催化氧化的高活性物种[17,25]. 同时Mn1.5Co2.5Ce1催化剂中高价态的Mn和Co含量也较高,这或许是其催化活性反而高于Mn2Co2Ce1催化剂的原因之一. 对比Ce 3d数据可以发现,随着Mn/Co的减小,Ce3+的含量也逐渐减少,而更多的Ce3+被认为与催化剂表面的电子不平衡和氧空位形成有关,有利于NO的催化氧化[26]. 这或许是Mn3Co1Ce1催化剂在拥有最少含量的高价Mn和Co同时仍然具备较高催化活性的原因. 从表3可以看到,Mn2.5Co1.5Ce1、Mn2.5Co1.5Ce1和Mn1.5Co2.5Ce1之间的化学吸附氧Oad差异不大(<2%),说明对于制备的MnCoCeOx催化剂,Oad含量不是活性的决定性因素. 值得注意的是,Mn2Co2Ce1催化剂的化学吸附氧Oad含量(38.7%)却明显低于其他3组催化剂平均水平(~44.0%). 有研究表明,化学吸附氧物种(Oad)比晶格氧Ola更有利于吸附态NO的氧化转化[27]. 因此推测过低的化学吸附氧Oad可能是除较小的比表面积外导致其催化活性较差的又一原因. 上述XPS结果表明,Mn/Co比通过影响MnCoCeOx催化剂中各元素价态进而对其催化氧化NO能力造成一定影响.
-
一般来说,催化剂的表面酸性和氧化还原能力对于反应物分子的吸附,中间产物的转化以及最终产物的脱附都有着重要影响,因此对各MnCoCeOx催化剂进行了NH3-TPD和H2-TPR测试,NH3-TPD的结果如图6(a)所示. 以Mn2.5Co1.5Ce1催化剂为例,可将温度划分为低温区(50—350 ℃)和高温区(350—750 ℃). NH3-TPD曲线上低区内118 ℃和215 ℃的脱附峰可归因为吸附在催化剂弱酸位上NH3的脱附,而在高温度374 ℃和590 ℃的峰则是与强酸位结合的NH3脱附所致[28]. 对比表3中各催化剂相应酸位上NH3的脱附量,可以发现几组催化剂的NH3总脱附量大小顺序与催化活性结果对应一致,而强酸位上的NH3脱附量差异较小,NH3脱附量的差异主要集中在弱酸位上,这说明对于MnCoCeOx催化剂,可能弱酸位对其催化活性有着更大的影响.
图6(b)展示了各催化剂的H2-TPR测试结果,各催化剂的H2-TPR曲线上均出现了3个明显的还原峰. 其中,235 ℃和327 ℃(363 ℃)的低温还原峰对应于MnO2→Mn2O3→Mn3O4的还原过程[29]. 而563 ℃(540 ℃)的高温还原峰则对应CeO2→Ce2O3的还原过程[30],这一结果也与XRD和XPS分析所对应. 从可以看出,随着Mn/Co比的减小,各还原峰位置逐渐向低温移动,说明Co掺杂含量的增加有利于催化剂的氧化还原能力的增强. 而还原峰的面积可以反映催化剂的氧化还原能力大小,通过对还原峰面积进行积分计算,可以得到各还原峰对应的氢气消耗量,结果如表4所示. 可以看出,与活性组分Mn相关的还原峰(区域I)的耗氢量大小顺序为Mn2.5Co1.5Ce1 > Mn3Co1Ce1 > Mn1.5Co2.5Ce1 > Mn2Co2Ce1,与活性顺序对应一致. 而随着Mn/Co比的减小,对应于Ce物种还原峰(区域Ⅱ)的耗氢量逐渐增大,说明催化剂中CeO2含量逐渐增大,这于XPS分析结果对应一致. 上述结果说明Mn2.5Co1.5Ce1催化剂有着更丰富的表面酸性位点和更强的氧化还原能力.
-
通过柠檬酸-溶胶凝胶法制备的Mn2.5Co1.5Ce1催化剂在368 mg·m−3 NO,10 % O2,体积空速
28000 h−1条件下,在260 ℃达到了83.9%的NO转化率,并在250—300 ℃的宽温度区间内保持80%以上转化率,具有良好的催化性能. 而当样品中Mn/Co比逐渐减小时,MnCoCeOx催化剂催化氧化NO的活性并未呈现出简单的变化规律,表明适当的Mn/Co比例才有利于提高的催化性能. 通过系列BET、XRD、XPS、NH3-TPD和H2-TPR表征结果表明,Mn/Co比对MnCoCeOx催化剂的孔隙结构、元素价态、表面酸性和氧化还原能力都有着明显的影响,进而对其催化性能产生较大影响. 其中,Mn:Co:Ce=2.5:1.5:1的Mn2.5Co1.5Ce1催化剂得益于更大的比表面积和孔容,更多的高价态Mn3+、Mn4+和Co3+物种和酸性位点,以及更强的氧化还原能力,具备了最佳的催化能力. 为实现催化烟气中氧气氧化NO,进一步提升臭氧多脱技术的经济性提供了新的思路.
锰钴铈复合氧化物催化氧化NO性能研究
Study on the catalytic oxidation of NO over manganese cobalt cerium composite oxides
-
摘要: 通过柠檬酸溶胶凝胶法成功制备了系列MnCoCeOx催化剂,用于催化烟气中残留氧气氧化NO. XRD、BET、XPS、NH3-TPD和H2-TPR等表征结果表明Mn2.5Co1.5Ce1Ox催化剂比表面积、孔容更大,具有更多的表面高价态Mn3+、Mn4+、Co3+物种,丰富的表面酸性位点以及更强的低温氧化还原能力,有利于NO的催化氧化. Mn2.5Co1.5Ce1Ox催化剂能在200—350 ℃的宽温度范围内保持60%以上的NO氧化效率,并且在260 ℃下达到最大值83.9%,为催化烟气中残留氧气氧化NO提供了新思路.
-
关键词:
- MnCoCeOx复合氧化物 /
- 催化氧化 /
- NO /
- 酸性位.
Abstract: A series of MnCoCeOx catalysts were successfully prepared by a citric acid sol-gel method and used for catalyzing the oxidation of NO by residual oxygen in the flue gas. The characterization results of XRD, BET, XPS, NH3-TPD, and H2-TPR implied that Mn2.5Co1.5Ce1Ox catalyst had a larger surface area and pore volume, more surface high-valence Mn3+, Mn4+, Co3+ species, abundant surface acidic sites and stronger low-temperature redox ability, which were conducive to the catalytic oxidation of NO. Mn2.5Co1.5Ce1Ox catalyst could maintain a NO oxidation efficiency of over 60% in a wide temperature range of 200—350 ℃, and reached a maximum of 83.9% at 260 ℃, providing a novel idea for catalytic oxidation of NO with residual oxygen in the flue gas.-
Key words:
- MnCoCeOx /
- catalytic oxidation /
- NO /
- acidic site.
-

-
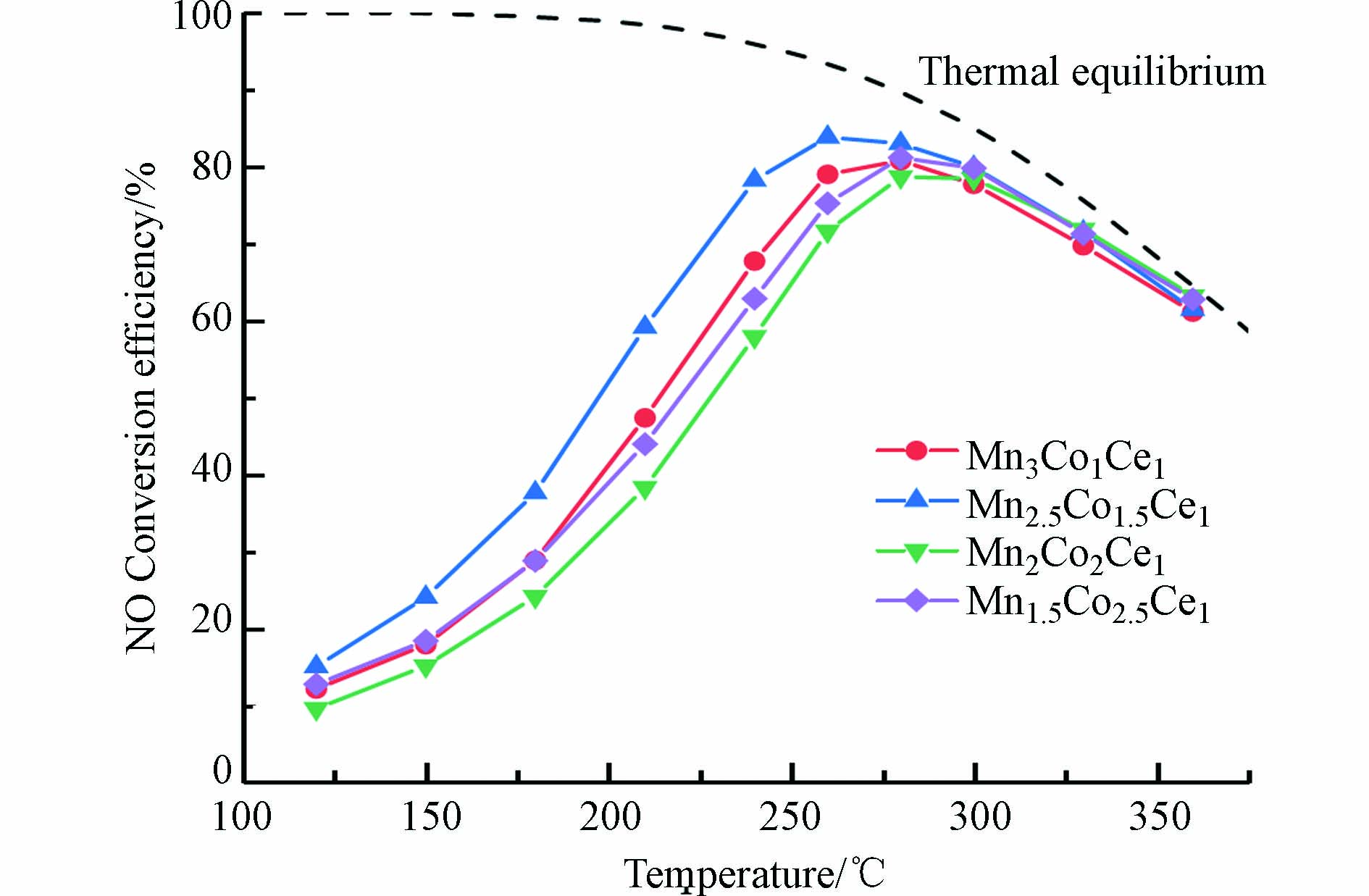
图 1 不同MnCoCeOx催化剂的NO转化效率
Figure 1. NO conversion efficiency of different MnCoCeOx catalysts

图 4 不同MnCoCeOx催化剂N2吸脱附曲线和孔径分布
Figure 4. Catalytic N2 absorption and desorption curves and pore size distribution of different MnCoCeOx catalysts

图 5 不同MnCoCeOx催化剂的XPS图谱(a) Ce 3d; (b) Mn 2p; (c) Co 2p; (d) O 1s
Figure 5. XPS spectra of different MnCoCeOx catalysts

图 6 不同MnCoCeOx催化剂NH3-TPD(a)和H2-TPR(b)图谱
Figure 6. NH3-TPD (a) and H2-TPR (b) results of different MnCoCeOx catalysts
表 1 本文制备的 MnCoCeOx 催化剂与文献报道的 Mn 基催化剂催化氧化 NO 的性能对比
Table 1. The comparison of NO oxidation efficiency among the reported Mn-based catalysts and thesynthesized MnCoCeOx catalyst in this work
 下载: 导出CSV
下载: 导出CSV
表 2 不同MnCoCeOx催化剂孔结构参数
Table 2. Pore structure parameters of different MnCoCeOx catalysts
催化剂
Catalysts比表面积/( m2·g−1)
Specific surface area孔容a/(cm3·g−1)
Pore volume平均孔径b/nm
Average Pore diameterMn3Co1Ce1 88.94 0.101 5.90 Mn2.5Co1.5Ce1 108.37 0.096 5.50 Mn2Co2Ce1 60.80 0.075 6.88 Mn1.5Co2.5Ce1 67.72 0.085 6.72 a BJH 脱附孔总体积,b BJH 脱附平均孔径
下载: 导出CSV
表 3 不同MnCoCeOx催化剂的XPS数据
Table 3. XPS data of different MnCoCeOx catalysts
催化剂
CatalystsMn 2p Ce 3d Co 2p O 1s Mn2+/ % Mn3+/ % Mn4+/ % Ce3+/ % Ce4+/ % Co2+/ % Co3+/ % Oad/ % Ola/ % Mn3Co1Ce1 19.42 49.69 30.89 24.69 75.31 44.24 55.76 44.77 55.23 Mn2.5Co1.5Ce1 12.47 53.55 33.97 22.77 77.23 35.65 64.35 43.72 56.28 Mn2Co2Ce1 17.48 49.28 33.24 21.65 78.35 57.98 42.02 38.70 61.30 Mn1.5Co2.5Ce1 13.39 51.69 34.92 18.27 81.73 38.36 61.64 46.14 53.86
下载: 导出CSV
表 4 不同MnCoCeOx催化剂氢气消耗量
Table 4. The H2 consumption of different MnCoCeOx catalysts
催化剂
Catalysts区域Ⅰ耗氢量/(mmol·g−1)
Region Ⅰ H2 consumption区域Ⅱ耗氢量/(mmol·g−1)
Region Ⅱ H2 consumption总耗氢量/(mmol·g−1)
Total H2 consumptionMn3Co1Ce1 2.79 0.55 3.34 Mn2.5Co1.5Ce1 2.88 1.05 3.93 Mn2Co2Ce1 2.59 1.69 4.28 Mn1.5Co2.5Ce1 2.65 2.56 5.21
下载: 导出CSV
-
[1] 陈翠芝, 陈伟国. 城市主要大气污染物与呼吸系统疾病相关性浅析[J]. 上海环境科学, 1994(9): 27-30,46. CHEN C Z, CHEN W G. Preliminary analysis of correlation between respiratory disease and urban air pollutants[J]. Shanghai Environmental Sciences, 1994(9): 27-30,46 (in Chinese).
[2] HONG Z, WANG Z, LI X B. Catalytic oxidation of nitric oxide (NO) over different catalysts: An overview[J]. Catalysis Science & Technology, 2017, 7(16): 3440-3452. [3] MA Q, WANG Z H, LIN F W, et al. Characteristics of O3 oxidation for simultaneous desulfurization and denitration with limestone–gypsum wet scrubbing: Application in a carbon black drying kiln furnace[J]. Energy & Fuels, 2016, 30(3): 2302-2308. [4] 苏士焜, 宗保宁, 荣峻峰. 金属氧化物催化剂用于NO催化氧化的研究进展[J]. 化工环保, 2022, 42(3): 249-254 doi: 10.3969/j.issn.1006-1878.2022.03.002 SU S K, ZONG B N, RONG J F. Research progress of metal oxide catalysts for catalytic oxidation of NO[J]. Environmental Protection of Chemical Industry, 2022, 42(3): 249-254(in Chinese) doi: 10.3969/j.issn.1006-1878.2022.03.002
[5] 王先涛, 马宏燎, 柏子龙. 锰铈复合氧化物用于NO催化氧化的研究[J]. 现代化工, 2017, 37(2): 92-95. WANG X T, MA H L, BAI Z. Catalytic oxidation of NO to NO2 on Mn-Ce-Ox catalyst[J]. Modern Chemical Industry, 2017, 37(2): 92-95 (in Chinese).
[6] GAO F Y, CHU C, ZHU W J, et al. High-efficiency catalytic oxidation of nitric oxide over spherical MnCo spinel catalyst at low temperature[J]. Applied Surface Science, 2019, 479: 548-556. doi: 10.1016/j.apsusc.2019.02.116 [7] 曹禄彬, 赵锋, 胡国新. Mn-Fe/γ-Al2O3的制备与NO催化氧化性能[J]. 实验室研究与探索, 2010, 29(8): 4-7. doi: 10.3969/j.issn.1006-7167.2010.08.002 CAO L B, ZHAO F, HU G X. Synthesis of Mn-Fe/γ-Al2O3 and its performance in catalytic oxidation of NO[J]. Research and Exploration in Laboratory, 2010, 29(8): 4-7 (in Chinese). doi: 10.3969/j.issn.1006-7167.2010.08.002
[8] LI K, TANG X L, YI H H, et al. Catalytic oxidation of NO over Mn-Co-Ce-O x catalysts: Effect of reaction conditions[J]. Research on Chemical Intermediates, 2014, 40(1): 169-177. doi: 10.1007/s11164-012-0953-7 [9] LI K, TANG X L, YI H H, et al. Low-temperature catalytic oxidation of NO over Mn-Co-Ce-O x catalyst[J]. Chemical Engineering Journal, 2012, 192: 99-104. doi: 10.1016/j.cej.2012.03.087 [10] JIANG B Q, WU Z B, LIU Y, et al. DRIFT study of the SO2 effect on low-temperature SCR reaction over Fe–Mn/TiO2[J]. The Journal of Physical Chemistry C, 2010, 114(11): 4961-4965. doi: 10.1021/jp907783g [11] LIN F W, SHAO J M, TANG H R, et al. Enhancement of NO oxidation activity and SO2 resistance over LaMnO3+ δ perovskites catalysts with metal substitution and acid treatment[J]. Applied Surface Science, 2019, 479: 234-246. doi: 10.1016/j.apsusc.2019.02.104 [12] ZHAO B H, RAN R, WU X D, et al. Phase structures, morphologies, and NO catalytic oxidation activities of single-phase MnO2 catalysts[J]. Applied Catalysis A:General, 2016, 514: 24-34. doi: 10.1016/j.apcata.2016.01.005 [13] LI M J, HUANG K, SCHOTT J A, et al. Effect of metal oxides modification on CO2 adsorption performance over mesoporous carbon[J]. Microporous and Mesoporous Materials, 2017, 249: 34-41. doi: 10.1016/j.micromeso.2017.04.033 [14] WANG X B, DUAN R B, LIU W, et al. The insight into the role of CeO2 in improving low-temperature catalytic performance and SO2 tolerance of MnCoCeO x microflowers for the NH3-SCR of NO x[J]. Applied Surface Science, 2020, 510: 145517. doi: 10.1016/j.apsusc.2020.145517 [15] 张弛. 多级结构钴基催化剂的制备及其净化VOCs机制研究[D]. 上海: 上海工程技术大学, 19-32. ZHANG C. Preparation of multi-stage cobalt-based catalyst and study on its mechanism of VOCs purification[D]. Shanghai: Shanghai University of Engineering Science: 19-32(in Chinese).
[16] MERCIER F, ALLIOT C, BION L, et al. XPS study of Eu(Ⅲ) coordination compounds: Core levels binding energies in solid mixed-oxo-compounds Eu mX xO y[J]. Journal of Electron Spectroscopy and Related Phenomena, 2006, 150(1): 21-26. doi: 10.1016/j.elspec.2005.08.003 [17] GUO Y H, WEI H Y, ZHAO G Y, et al. Low temperature catalytic performance of coal-fired flue gas oxidation over Mn-Co-Ce-Ox[J]. Fuel, 2017, 206: 318-324. doi: 10.1016/j.fuel.2017.06.034 [18] CHEN X P, LIU Q, WU Q, et al. A hollow structure WO3@CeO2 catalyst for NH3-SCR of NO x[J]. Catalysis Communications, 2021, 149: 106252. doi: 10.1016/j.catcom.2020.106252 [19] TANG X L, CHEN D, CHU C, et al. Mn-Co binary oxides for low-temperature catalytic oxidation of NO: Effect of SO2 and regeneration[J]. Journal of Chemical Technology & Biotechnology, 2021, 96(10): 2956-2964. [20] SHEN B X, ZHU S W, ZHANG X, et al. Simultaneous removal of NO and Hg0 using Fe and Co co-doped Mn-Ce/TiO2 catalysts[J]. Fuel, 2018, 224: 241-249. doi: 10.1016/j.fuel.2018.03.080 [21] GAO F Y, TANG X L, YI H H, et al. Novel Co–or Ni–Mn binary oxide catalysts with hydroxyl groups for NH3–SCR of NO x at low temperature[J]. Applied Surface Science, 2018, 443: 103-113. doi: 10.1016/j.apsusc.2018.02.151 [22] WARANG T, PATEL N, SANTINI A, et al. Pulsed laser deposition of Co3O4 nanoparticles assembled coating: Role of substrate temperature to tailor disordered to crystalline phase and related photocatalytic activity in degradation of methylene blue[J]. Applied Catalysis A:General, 2012, 423/424: 21-27. doi: 10.1016/j.apcata.2012.02.037 [23] TANG X L, GAO F Y, XIANG Y, et al. Effect of potassium-precursor promoters on catalytic oxidation activity of Mn-CoOx catalysts for NO removal[J]. Industrial & Engineering Chemistry Research, 2015, 54(37): 9116-9123. [24] SHAO J M, LIN F W, WANG Z H, et al. Low temperature catalytic ozonation of toluene in flue gas over Mn-based catalysts: Effect of support property and SO2/water vapor addition[J]. Applied Catalysis B:Environmental, 2020, 266: 118662. doi: 10.1016/j.apcatb.2020.118662 [25] WANG D D, CHENG J, WANG B H, et al. Plasma-catalytic high-efficiency oxidation of NO over Co-Mn/Ti catalysts using surface dielectric barrier discharge plasma[J]. Vacuum, 2019, 167: 249-254. doi: 10.1016/j.vacuum.2019.06.004 [26] GU T T, LIU Y, WENG X L, et al. The enhanced performance of ceria with surface sulfation for selective catalytic reduction of NO by NH3[J]. Catalysis Communications, 2010, 12(4): 310-313. doi: 10.1016/j.catcom.2010.10.003 [27] XIE C Y, ZHU B Z, SUN Y L, et al. Effect of doping Cr on NH3 adsorption and NO oxidation over the Fe xO y/AC surface: A DFT-D study[J]. Journal of Hazardous Materials, 2021, 416: 125798. doi: 10.1016/j.jhazmat.2021.125798 [28] CHEN G Y, WANG Z, LIN F W, et al. Comparative investigation on catalytic ozonation of VOCs in different types over supported MnO x catalysts[J]. Journal of Hazardous Materials, 2020, 391: 122218. doi: 10.1016/j.jhazmat.2020.122218 [29] PICASSO G, GUTIÉRREZ M, PINA M P, et al. Preparation and characterization of Ce-Zr and Ce-Mn based oxides for n-hexane combustion: Application to catalytic membrane reactors[J]. Chemical Engineering Journal, 2007, 126(2/3): 119-130. [30] LIANG Y L, HUANG Y F, ZHANG H L, et al. Interactional effect of cerium and manganese on NO catalytic oxidation[J]. Environmental Science and Pollution Research, 2017, 24(10): 9314-9324. doi: 10.1007/s11356-017-8645-x -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1164
- HTML全文浏览数: 1164
- PDF下载数: 83
- 施引文献: 0
