

-
随着城市化和工业化的快速发展,越来越多的挥发性有机化合物(volatile organic compounds,VOCs)被排放到大气中,这不仅会造成严重的环境污染,还会危害人体健康[1]. 在众多处理技术中,催化燃烧技术具有能耗低、处理效率高、无二次污染等优点,是一项拥有巨大发展潜力的技术. 催化燃烧技术的核心是选择合适的催化剂. 常见的催化剂有负载型贵金属催化剂[2 − 4]和过渡金属氧化物催化剂[5]. 其中过渡金属氧化物催化剂成本低廉,但催化性能相对较差;负载型贵金属催化剂具有催化性能好,使用寿命长等特点而受到广泛关注. 在甲苯催化氧化反应的研究中,Pt 基催化剂表现出良好的催化活性,其性能与金属与载体间的相互作用[2]、表面 Pt0 的含量[3]、贵金属分散度[4]等因素相关. 与单金属催化剂相比,双金属催化剂由于加入另一种金属会改变原来贵金属的电子和几何结构,具有协同效应,可以表现出更好的催化性能[6]. 例如,在Pt中引入Ir,贵金属 Ir 作为铂族金属之一,可以活化 C—H 键,并且能调节催化剂的电子结构进而提升催化剂的催化性能[7],因此,PtIr双金属催化剂的氧化性能优于 Pt 单金属催化剂.
除了催化剂的活性组分,不同制备方法,也会影响催化剂的物理化学性质,进而影响催化剂的催化性能. Zeng 等[8]采用浸渍法、沉积沉淀法和光沉积法合成了3种 Pt/TiO2 催化剂,并探究其对甲苯催化氧化活性的影响. 其中光沉积法所制备的 PtTi-P 催化剂由于具有最强的氧活化能力从而表现出最好催化活性. Sun等[9]研究发现,溶胶沉淀法制备的 IrAl-CD 催化剂较沉积沉淀法所制备的 IrAl-DP 催化剂具有更高含量的 Ir0 物种,因此在甲醛的催化氧化中表现出更优异的催化性能. Hu等[10]研究发现,制备方法对催化剂表面结构形貌产生显著影响,柠檬酸配合法制备的 CuMnCeOx 催化剂较水热法制备的催化剂具有更丰富的微孔结构,从而有利于对气态 HCHO 的吸附及氧化. 由此可见,催化剂制备方法的不同会使金属-载体间相互作用、催化剂中贵金属价态、催化剂表面结构形貌等催化剂物理化学性质发生改变,最终影响催化剂催化性能. 因此本文考察了制备方法对 Pt-Ir/CeO2 双金属催化剂催化氧化甲苯性能的影响并初步探讨其作用机理.
本文采用浸渍法、沉积沉淀法、乙二醇还原法制备了3种 Pt-Ir/CeO2 催化剂,以甲苯氧化为探针反应,探究不同制备方法对催化剂催化氧化甲苯性能的影响,并通过 XRF、HRTEM、EDS、BET、XRD、XPS、CO-DRIFTS、H2-TPR、O2-TPD、In-situ DRIFTS 等表征手段对所制备的催化剂进行物理化学性质表征,探讨其间存在活性差异的内在原因.
-
(1)浸渍法:先将商业购买的 CeO2 置于马弗炉中在 550 ℃ 焙烧 5 h,取 1.936 g 焙烧后的 CeO2 置于去离子水中搅拌,向其中加入 1.5 mL共 0.03 mmol IrCl3 溶液和 1.5 mL 共 0.03 mmol H2PtCl6·6H2O 溶液,继续搅拌 30 min,然后水浴 80 ℃ 蒸干. 将所得产物在 110 ℃ 下干燥 4 h,冷却研磨后用马弗炉 400 ℃ 煅烧 2 h. 随后,用氢气 400 ℃ 还原 2 h,得到样品 Pt-Ir/CeO2-IMP.
(2)沉积沉淀法:称取与上述相同量的 CeO2 置于去离子水中搅拌,并向其中加入相同量的 IrCl3 和 H2PtCl6·6H2O. 在搅拌过程中加 NaOH 溶液(0.1 mol·L−1),将混合溶液的 pH 调节至 10. 搅拌结束后洗涤离心,在 110 ℃ 下干燥 4 h,冷却研磨后在马弗炉中 400 ℃ 下煅烧 2 h,再用氢气 400 ℃ 还原 2 h. 得到样品 Pt-Ir/CeO2-DP.
(3)乙二醇还原法:取 15 mL 乙二醇溶液、100 mg 聚乙烯吡咯烷酮(polyvinylpyrrolidone,PVP) 加入三颈烧瓶,将其置于微波反应器中,打开磁力搅拌器. 升温至 150 ℃ 后,向三颈烧瓶中逐滴滴加与上述相同量的 IrCl3 和 H2PtCl6·6H2O的乙二醇溶液. 随后,逐滴滴加 2.4 mL NaOH 溶液(0.25 mol·L−1),反应 30 min. 反应结束后,将烧瓶置于冰水浴 30 min. 所得混浊液先用丙酮进行洗涤离心,后用乙醇和正己烷的混合溶液洗涤离心. 得到的产物分散在乙醇溶液中,并将其滴加在预先分散于乙醇中的 CeO2 溶液中,搅拌后水浴蒸干,在 110 ℃ 下干燥 4 h. 得到样品 Pt-Ir/CeO2-EG.
-
以甲苯氧化为探针反应,评价催化剂的催化氧化 VOCs 的性能. 反应在自组装气相固定床微型反应器上进行. 取 0.1 g 经压片过筛的 20—40 目的催化剂置于反应管中部并以石英棉固定,将反应管置于可程序升温的反应炉中. 采用鼓泡法将甲苯在 25 ℃ 水浴条件下鼓入混合缓冲瓶中,经缓冲瓶充分混匀后的气体进入反应管,反应尾气进入气相色谱仪(GC-
9860 )进行在线检测分析,气相色谱配Porapak Q色谱柱、火焰离子化检测器(FID)和热导检测器(TCD). 本文采用的甲苯浓度为 0.1%,空速(GHSV)为30000 mL· g−1 ·h−1,燃烧曲线是在每个反应温度点停留30 min,每个点采集3次平行样,以平均值作为活性结果.甲苯转化率(Xtoluene,%)定义如公式(1)所示:
CO2 产率(
$Y_{{\mathrm{CO}}_2} $ ,%)是衡量甲苯氧化程度的重要指标,通过公式(2)计算获得:其中,Cin 和 Cout 分别为进样气中入口和出口处的甲苯浓度.
$C_{{\mathrm{CO}}_2} $ 代表出气气体中 CO2 浓度. -
催化剂活化能(Ea)值的计算条件为甲苯转化率低于 20 % 的温度区间,通过 Arrhenius 公式计算获得,公式如(3)所示:
其中,k 为反应的速率常数;A 为指前因子;R 为摩尔气体常数;T 为反应温度.
-
使用 Philips X'Pert Pro 衍射仪采集样品的 X 射线衍射谱图(XRD). 激发光源为Cu Kα 辐射(λ=
0.15418 nm),采用仪器的工作电压为 40 kV,电流为 40 mA,扫描速度为 10 (°)·min−1. 使用 ARL9800XP+ 型 X 射线荧光光谱仪对所制备样品中贵金属 Pt 和 Ir 的实际含量进行分析,确定Pt、Ir 比例. 使用 Micrometrics ASAP 2020 吸附仪测定 N2 吸-脱附曲线,用 Brunauer Emmett-Teller (BET)法和 Barret-Joyner-Halenda(BJH)法获得样品的比表面积和孔径分布. 使用 JEM-2100 (电压为 200 kV)仪器收集样品的 HRTEM 图,观察样品的形貌及晶格间距. 使用 JEM-2010 LaB6 型高分辨透射电子显微镜对样品进行观察,利用高角度环形暗场扫描透射电子显微镜(HAADF STEM)和 X 射线能谱(EDS)研究样品的形貌和元素分散状态. 使用 PHI5000 Versa Probe 型X射线光电子能谱仪搜集样品元素的 X 射线光电子能谱(XPS) ,激发光源为Al Kα 辐射(1486.6 eV)单色散射线,所得到的结合能均采用 C 1s(284.6 eV)作为标准进行校正. 使用 Nicolet iS50 FTIR 光谱仪进行了 CO 化学吸附原位红外光谱测试(CO-DRIFTS),搜集样品表面贵金属的存在状态. 测试条件如下:首先通入 25 mL·min−1 N2 在 200 ℃ 下对样品吹扫 1 h,冷却至室温后采集背景谱图. 然后将 N2 切换为 1% CO/N2. 吸附 50 min(吸附饱和)后,再切换为 N2,吹扫 50 min,记录 CO 吸附红外光谱图. 使用 Nicolet iS50 FTIR 光谱仪对样品进行了甲苯原位漫反射傅里叶变换红外光谱(In-situ DRIFTS)测试,研究甲苯在样品上的反应过程. 测试条件如下:使用 N2 气(25 mL·min−1)在 200 ℃ 下对样品吹扫 60 min,然后在冷却过程中采集所需温度下的背景谱图,直至温度冷却到室温时,使用 0.1% 甲苯/空气代替 N2 通入装置,吸附饱和后开始升温,在所需温度下采集原位红外谱图. 使用 Finesorb-3010 型化学吸附仪进行 H2 程序升温还原来研究样品的还原性能,检测器为 TCD 检测器. 测试条件如下:称量约 10 mg 样品,将其在高纯 N2 气氛下 200 ℃ 吹扫 1 h,随后,冷却至室温,通入 7% H2/Ar. 待其吸附饱和后将样品加热至 700 ℃,升温速率为 10 ℃·min−1,记录该过程 H2 消耗量. 使用 ASAP2920 程序升温化学吸附仪进行 O2 程序升温脱附测试研究样品氧物种存在状态. 测试条件如下:取 100 mg 样品在 He 气氛下 200 ℃ 吹扫 1 h,冷却至室温后,通入 40 mL·min−1 O2 吸附 30 min,然后再通入 He 气进行吹扫 30 min,最后升温进行脱附,以 10 ℃·min−1 的升温速率升温至 800 ℃,记录该过程 O2 的脱附量. -
首先,通过 XRF 表征以获得不同方法所制备的双金属 Pt-Ir/CeO2 样品中贵金属 Pt 和 Ir 的实际含量,结果如表1所示. 结果表明,Pt-Ir/CeO2-IMP 样品中贵金属 Pt 和 Ir 含量的实际值与理论值接近,Pt、Ir贵金属含量基本都为 0.3%. 而 Pt-Ir/CeO2-DP、Pt-Ir/CeO2-EG 样品中贵金属 Pt 和 Ir 含量的实际值偏小,这可能是由于在洗涤离心过程中造成了一定量的损失. 但3种方法所制备的双金属 Pt-Ir/CeO2 样品中总贵金属载量相差较小,并且3种样品中 Pt/Ir 物质的量比相接近,在 1.0—1.2 的范围内,因此具有可比性.
对3种不同方法所制备的双金属 Pt-Ir/CeO2 样品进行了甲苯催化氧化活性测试和 CO2 产率测试,结果如图1(a)和1(b)所示. 不难发现,随着反应温度的升高,3种催化剂的甲苯转化率均逐渐提升,并且在低温段转化率升高的较缓慢,在高温处提升的较快. 并且发现 CO2 产率与甲苯催化活性结果相似,这说明3种催化剂选择性都较好. 此外,T10、T50、T90 分别对应甲苯转化率分别为 10%、50%、90% 的反应温度,常用于比较样品间反应活性差异,因此将其列于表2. 由表2可知,Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 3种样品 T10 值分别为 136 ℃、122 ℃ 和 122 ℃,T90 值分别为 237 ℃、223 ℃ 和 163 ℃. 特征温度越高,则反应活性越差. 结合上述结果,不难发现,Pt-Ir/CeO2-EG 样品 T90 值最小,表现出最优异的催化氧化甲苯性能,在 180 ℃ 条件下就能完全转化甲苯;Pt-Ir/CeO2-DP次之;而 Pt-Ir/CeO2-IMP 甲苯催化氧化活性最差.
为了研究催化剂的稳定性,在甲苯浓度为 0.1% ,GHSV 为
30000 mL·g−1·h−1,反应温度为 165 ℃ 的条件下,对 Pt-Ir/CeO2-EG 催化剂进行稳定性评估试验,结果如图1(c)所示. 可以发现,在 24 h 的稳定性实验中,活性始终保持在 90% 左右,说明催化剂具有良好的热稳定性.为了研究3种催化剂对于催化反应的活化能差异,通过动力学计算相关数据,其表观活化能(Ea)由Arrhenius 曲线斜率确定,结果如图1(d)所示,并记录在表2中. 结果表明,3种催化剂的活化能值顺序与甲苯转化率结果较好吻合. Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-IMP 催化剂的 Ea 值相差并不明显,但 Pt-Ir/CeO2-EG 催化剂的 Ea 值远低于这两种催化剂,仅有 35.88 kJ·mol−1,这说明了 Pt-Ir/CeO2-EG 催化剂上更容易活化氧化甲苯.
-
对3种样品和载体进行 XRD 分析以研究样品的晶体结构,结果如图2所示. 从图2可以看出,Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 3种样品的图谱与 CeO2 载体的图谱几乎一致,在28.55°、33.08°、47.48°、56.33°、59.08°、69.40°、76.70°、79.07° 和 88.41° 的位置处出现衍射峰,这些衍射峰分别对应 CeO2(JCPDS PDF# 34-0394)的(111)、(200)、(220)、(311)、(222)、(400)、(311)、(420)、(422)晶面,表明负载双金属没有破坏 CeO2 的立方萤石结构[3]. 虽然 XRF 结果证明了 Pt、Ir 物种的存在,但在 XRD 光谱中并未发现贵金属 Pt和 Ir 有关的特征衍射峰,这说明 Pt、Ir 在 CeO2 表面均匀分散,或 Pt、Ir 载量较低,低于 XRD 的检出限.
对样品及载体进行 N2 吸-脱附等温线分析,以获得样品的比表面积与孔径分布信息,结果如图3和表3所示. 由图3吸附等温线可以看出,3种方法制备的样品以及载体均属于具有 H3 型回滞环的 Ⅳ 型吸附等温线. 比表面积按照 CeO2 > Pt-Ir/CeO2-IMP > Pt-Ir/CeO2-DP > Pt-Ir/CeO2-EG 的顺序分别为65.1、64.7、63.6、59.4 m2·g−1. 从 BJH 孔径分布曲线图中可以看出,催化剂和载体的孔径呈规则的单峰分布,且孔径相似,均在 15—16 nm 范围内. 与载体 CeO2 相比,负载 Pt、Ir 后可能导致堵塞载体中部分孔隙,从而造成样品的比表面积略微减小.
对样品进行 HRTEM 和 EDS 表征,以探究不同方法制备的 Pt-Ir/CeO2 样品的形貌与表面元素分布状态的差异,结果如图4所示. 其中图4(a)、4(c)以及4(e)分别为 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 样品的 HRTEM 图,对其晶格条纹进行测量,3种样品均观察到 0.320 nm 的晶格条纹,归属为二氧化铈的(111)晶面[11]. 此外,在图4(a)和图4(c)中,Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 两种样品并没有发现 PtIr 粒子,可能是因为 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 两种样品负载的贵金属粒径较小,而在图4(e)中 Pt-Ir/CeO2-EG 样品可以明显发现 PtIr 粒子,说明Pt-Ir/CeO2-EG 样品负载的贵金属粒径较大. 从图4(b),4(d),4(f)的 EDS 谱图中可以看到 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP、Pt-Ir/CeO2-EG 的3种样品负载的 Pt、Ir 粒子均匀分散在载体表面. 因此可以认为不同方法制得的样品 Pt 和 Ir 粒子都在 CeO2 表面均匀分散,这与 XRD 图谱的结论一致.
-
样品表面贵金属状态不同会对催化剂催化氧化甲苯性能产生影响. 为了解不同方法所制备的样品中表面双金属 Ir 和 Pt 的化学状态和相对含量,对样品进行 XPS 表征,结果如图5所示. 图5(a)为3种样品 Ir 4f 的 XPS 谱图,Ir 4f 由 Ir 4f7/2 和 Ir 4f5/2 组成,其中结合能位于 60.7 eV 和 63.9 eV 处的峰归属于 Ir0 物种;结合能位于 61.5 eV 和 64.9 eV 的峰归属于 Irδ+ 物种[9]. 结果表明,3种样品均存在金属态和氧化态的 Ir 物种,但3种样品金属态 Ir0 、氧化态 Irδ+ 所占比例并不相同,因此计算3种样品中 Ir 的不同价态的相对含量,结果如表4所示. 可以看到,Pt-Ir/CeO2-EG 样品中金属态 Ir0 相对含量最高. 查阅文献可知,催化剂中金属态 Ir0 物种可以向 O2 的反键 π* 轨道提供电子,因此 Ir0 的存在有助于对 O2 的活化,从而促进甲苯催化氧化反应的进行[12]. 图5(b)为3种样品 Pt 4f 的 XPS 谱图,Pt 4f 由 Pt 4f7/2 和 Pt 4f5/2 组成,其中结合能位于 71.0 eV 和 74.4 eV 的峰归属于 Pt0 物种;结合能位于 72.0 eV 和 75.4 eV 的峰归属于 Pt2+ 物种;结合能位于 74.0 eV 和 77.2 eV 的峰归属于 Pt4+ 物种[4]. 3种样品的 Pt 不同价态的相对含量如表4 所示,Pt-Ir/CeO2-EG 样品中 Pt0 相对含量最高,氧化态 Ptδ+ (Pt2++ Pt4+)最少;而Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 样品则主要存在氧化态 Ptδ+. 有文献认为 Pt0 物种有助于甲苯催化氧化反应的进行,通常被认为是活性位点[13]. 因此,Pt0 物种含量越高,甲苯催化氧化反应活性越好. 基于上述结果,Pt0、Ir0 物种比例大小的顺序为 Pt-Ir/CeO2-EG > Pt-Ir/CeO2-DP > Pt-Ir/CeO2-IMP,与反应活性顺序相一致,表明金属态物种对甲苯氧化贡献较大.
此外,氧空位可以吸附活化氧气,从而促进反应的进行,是影响催化氧化甲苯性能重要因素之一[14]. 因此通过Ce 3d和O 1s的XPS谱图对3种样品表面的氧空位进行分析. 图6(a)为样品 Ce 3d 的 XPS 图谱,对其进行分峰,结合能位于 916.4 eV (u"')、907.1 eV (u")、900.7 eV (u)、898.1 eV (v"')、888.5 eV (v")和 882.1 eV (v)的峰归属于表面 Ce4+ 物种,903.2 eV (u')和 884.7 eV (v')归属于表面 Ce3+ 物种[13]. 从图中可以发现,3种样品均存在 Ce3+ 和 Ce4+ 物种,根据峰面积比例计算得到各样品的 Ce3+/(Ce3++Ce4+) 量,如表4所示. 其中,Pt-Ir/CeO2-EG 样品的 Ce3+ 含量最高. 有文献表明,Ce3+ 的形成有利于表面氧空位的形成[14]. 因此可以认为 Pt-Ir/CeO2-EG 样品较 Pt-Ir/CeO2-DP、Pt-Ir/CeO2-IMP 样品氧空位浓度更高,这有利于甲苯催化氧化反应的进行. 图6(b)为样品 O 1s 的 XPS 图谱,其中位于 529.1 eV 的峰归属于表面晶格氧(Olatt);位于 531.6 eV 的峰归属于表面吸附氧(Oads);位于 533.1 eV 的峰归属于吸附的羟基或水分子氧(O-OH)[6]. 查阅文献可知,表面吸附氧(Oads)是甲苯催化氧化过程中重要活性物种[2]. 计算3种样品中 Oads 的含量,并列于表4 中. 样品 Oads 含量大小顺序为:Pt-Ir/CeO2-EG > Pt-Ir/CeO2-IMP > Pt-Ir/CeO2-DP,这与 Ce3+ 含量结果相一致.
对样品进行 CO 吸附原位红外表征,进一步探究样品表面贵金属 Pt、Ir 物种存在状态,结果如图7所示. 不难发现,不同方法制备样品的 CO 吸附振动峰的波数区间并不相同. 查阅文献可知,位于
2103 cm−1 和2085 cm−1 处的振动峰分别归属为 CO 在 Pt2+ 和 Ptδ+ 上的线性吸附[15],并且这两处的振动峰也可归属为 CO 在 Irδ+ 上的线性吸附[16]. 而位于2072 cm−1 和 2050 cm−1 处的振动峰归属为 Pt0-CO、Ir0-CO 线性吸附[8, 17]. 从 CO-DRIFTS 图谱可知,Pt-Ir/CeO2-IMP 样品表面双金属 Pt、Ir 主要是以氧化态形式存在;Pt-Ir/CeO2-DP 样品表面金属以氧化态和金属态形式存在;Pt-Ir/CeO2-EG 样品表面金属则主要以金属态形式存在,这也印证了上述 Ir、Pt 的XPS 结果. -
对样品进行了 O2-TPD 表征,以比较各样品对氧气活化能力的差异,结果如图8(a)所示. 一般来说,吸附的氧通过以下过程发生变化:物理吸附氧 O2(ad)→化学吸附氧O2−(ad)→化学吸附氧O−(ad)→晶格氧O2−(lattice)[18]. 其中物理吸附氧 O2(ad)和化学吸附氧 O2−(ad)相对而言比较容易脱附,而晶格氧物种 O2−(lattice)较难脱附. 根据文献报道:120—220 ℃ 的脱附峰归属于化学吸附氧 O2−;270—400 ℃ 的脱附峰可以归属于化学吸附氧 O−,两种氧分子都属于表面吸附的氧物种,可以直接参与催化反应,属于活性氧物种[19]. 3种样品的脱附峰面积大小顺序为:Pt-Ir/CeO2-EG > Pt-Ir/CeO2-IMP > Pt-Ir/CeO2-DP,说明 Pt-Ir/CeO2-EG 样品具有最多的化学吸附氧物种. 此外,Pt-Ir/CeO2-EG 样品解吸温度低于Pt-Ir/CeO2-IMP 和 Pt-Ir/CeO2-DP 样品,表明其具有更好的活化氧能力. 结合 Ce 3d,O 1s XPS 结果,可以发现 Pt-Ir/CeO2-EG 样品具有更强的吸附和活化氧气的能力,这可能是其甲苯催化活性更为优异的原因之一. 但是, O2-TPD 图谱还显示,Pt-Ir/CeO2-IMP 样品较 Pt-Ir/CeO2-DP 样品具有更好的吸附氧和活化氧的能力,这与两种样品的甲苯催化氧化活性结果不相符,可能是因为表面氧空位浓度和化学吸附氧的含量在甲苯催化氧化反应中并不是决定性的因素,而催化剂表面双金属的化学状态在甲苯的催化氧化反应中是更为重要的影响因素.
H2-TPR常用于表征催化剂的氧化还原能力,因此,对样品进行 H2-TPR 表征,以探究Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 样品的氧化还原性质的差异,结果如图8(b) 所示. 图中 CeO2 样品在 403 ℃ 和 539 ℃ 处存在还原峰,分别归属为表面氧物种和表面 Ce4+ 的还原[20]. 与载体 CeO2 相比,3种样品负载贵金属 Pt 和 Ir 后,还原峰向低温移动,这表明贵金属Pt、Ir 的加入提升了 CeO2 的还原能力. 此外,在低于 200 ℃ 的低温段出现新的还原峰. 查阅文献可知,低于 100 ℃ 的还原峰归属为 PtOx,IrOx 和吸附氧物种的还原[1, 17];而在 100—200 ℃ 范围内的还原峰归属为 Pt 或 Ir 附近 CeO2 表面氧的还原[21 − 22]. Pt-Ir/CeO2-EG 样品相较于Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 样品,仅在 136 ℃便出现Pt 或 Ir 附近 CeO2 表面氧的还原峰,还原峰出现温度较其他样品更低,这可能是由于Pt-Ir/CeO2-EG 样品的Pt-Ce相互作用更强[23]. 由此可见,Pt-Ir/CeO2-EG 样品具有最佳的低温还原性,这有助于甲苯催化燃烧.
-
为探究制备方式对甲苯催化氧化反应机理的影响,对3种样品进行甲苯原位红外表征,图9显示了3种催化剂的原位红外光谱. 图9(a)和9(b)为 Pt-Ir/CeO2-IMP 样品在100—260 ℃ 的甲苯原位红外光谱图. 可以发现,在100 ℃ 时,
1589 cm−1和1490 cm−1 处检测到的振动峰归属于芳环的骨架振动[17];位于1177 cm−1和1024 cm−1 处的振动峰分别归属于芳环的不对称 C—C 伸缩振动和 C—H 弯曲振动,这些振动峰的出现表明甲苯吸附在催化剂表面. 温度升高至 120 ℃ 时,观察到在1149 、1098 、1068 cm−1 处出现归属于苯甲醇的ʋ (C—O) 振动吸收峰[24];位于 1557 cm−1和1542 cm−1 处的振动峰归属于苯甲酸的不对称 ʋ (COO) 伸缩振动[25];位于1699 cm−1和1671 cm−1 处的振动峰归属于苯甲醛的 C=O 不对称伸缩振动[26],表明在120 ℃ 下催化剂表面聚集了苯甲酸、苯甲醇和苯甲醛等中间产物. 此外,温度升至140 ℃ 后,在1732 、1498 、1435 、1396 、1368 、1360 cm−1 处观察到多个振动峰. 查阅文献可知,1360 、1368 、1396 cm−1 处出现的峰归属于较短羧酸盐物种的ʋ (COO) 振动[27];1435 、1498 cm−1 处的峰属于较短的马来酸盐[28],而1732 cm−1 处的峰表明马来酸酐的存在[29]. 这些振动峰的变化说明,在120 ℃ 下,甲苯先被部分氧化为苯甲醇、苯甲醛和苯甲酸等中间产物在催化剂表面积累,随着温度的升高至140 ℃,上述中间物种进一步氧化,苯环被破坏,生成了马来酸酐和羧酸盐等小分子有机物. 此外,图9(a)还可以看到,100 ℃在2360 cm−1和2324 cm-1 处出现了微弱的 CO2 的振动峰 [30],其峰强度随反应温度升高而增强,说明反应温度越高,甲苯氧化越彻底.图9(c)和9(d)为 Pt-Ir/CeO2-DP 样品在 100—260 ℃ 下的甲苯原位红外光谱图. 温度达到 100 ℃ 时,在
1150 、1098 、1069 、1559 、1542 、1698 、1669 cm−1 处便出现了归属于苯甲酸、苯甲醇和苯甲醛等中间产物的振动峰,相较于 Pt-Ir/CeO2-IMP 样品,温度降低了 20 ℃. 此外,温度升至 120 ℃ 后,出现了马来酸酐、羧酸盐等小分子有机物的振动峰,说明催化剂表面中间产物进一步氧化,中间产物开环后后生成马来酸酐、羧酸盐等小分子有机物. 较 Pt-Ir/CeO2-IMP 样品,温度降低了 20 ℃.图9(e)和9(f)为 Pt-Ir/CeO2-EG 样品在 80—180 ℃ 下的甲苯原位红外光谱图. 和Pt-Ir/CeO2-IMP 样品相比,在 80 ℃下,除了检测到苯环的骨架振动峰,还检测到归属于芳环的 C—H 伸缩振动峰(
3067 cm-1 和3027 cm−1 )[31],表明 Pt-Ir/CeO2-EG 样品表面吸附甲苯温度降低了 20 ℃. 同时,在该温度下,检测到苯甲醇、苯甲酸等中间产物振动峰;此外,还在2928 cm−1和2896 cm−1 处出现归属于亚甲基的对称和不对称伸缩振动的新峰和在1243 cm−1 处出现属于表面酚盐的 ʋ (C—O) 伸缩振动新峰[32 − 33],表明样品表面产生更多的中间产物. 当温度升高至 100 ℃ 时,除了出现马来酸酐、羧酸盐等小分子有机物的振动峰,在1724 cm−1 处还出现了归属于苯基环氧化生成了醛或酮的峰[34]. 可见,Pt-Ir/CeO2-EG 样品表面出现更丰富的中间体,并且相较于 Pt-Ir/CeO2-IMP 样品,甲苯开环温度降低了 40 ℃.基于上述结果,推测甲苯在 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和Pt-Ir/CeO2-EG 样品上表现出相似的反应路径:甲苯→苯甲醇→苯甲醛→苯甲酸→马来酸酐、丙酮、羧酸盐等小分子有机物→CO2 和 H2O. 查阅文献可知,苯环开环是甲苯催化氧化过程中一个重要的步骤[35]. 总结甲苯原位红外数据发现,3种样品表面吸附甲苯温度和甲苯氧化开环生成小分子有机物温度并不相同,其中 Pt-Ir/CeO2-EG 样品温度最低,在 80 ℃ 下就出现了芳环振动峰,相较于Pt-Ir/CeO2-IMP 样品表面吸附甲苯温度降低了 20 ℃;在 100 ℃ 下出现小分子有机物振动峰,相较于 Pt-Ir/CeO2-IMP 样品,甲苯开环温度降低了 40 ℃. 这说明 Pt-Ir/CeO2-EG 样品表现出优异的低温氧化活性. 这可能是由于 Pt-Ir/CeO2-EG 样品具有更多活性位点(Pt0)和更强氧活化能力(Ir0 和氧空位浓度).
-
本文采用浸渍法、沉积沉淀法以及乙二醇还原法制备了3种双金属 Pt-Ir/CeO2 催化剂,以甲苯作为探针污染物,评价了不同制备方法样品催化氧化甲苯的反应活性,并借助一系列表征手段对催化剂的结构性质、理化性质等进行测试分析. 结果表明,在贵金属 Pt、Ir 载量和比例接近的情况下,乙二醇还原法制备的 Pt-Ir/CeO2-EG 样品具有最佳的催化氧化甲苯活性,其 T90 为 163 ℃,远低于 Pt-Ir/CeO2-DP(223 ℃)和 Pt-Ir/CeO2-DP (246 ℃). HRTEM、EDS、XRD 表征说明,不同方法制备的样品中的贵金属 Pt 和 Ir 均匀分散在载体 CeO2 表面. XPS、CO-DRIFTS、H2-TPR、O2-TPD 表征说明,乙二醇还原法制备的 Pt-Ir/CeO2 样品具有最高比例的金属态贵金属 Pt 和 Ir,并且具有最多的化学吸附氧物种,吸附氧和活化氧的能力最强. In-situ DRIFTS表征说明,沉积沉淀法和乙二醇还原法制备的 Pt-Ir/CeO2 样品在 100 ℃ 条件下就可以破坏甲苯苯环将甲苯氧化为马来酸酐、丙酮、羧酸盐等小分子有机物,两种样品具有较好的低温活性. 因此,在Pt-Ir/CeO2 双金属催化剂中,金属态贵金属 Pt0 为活性位点,Ir0 的存在有助于促进甲苯催化氧化活性,其中乙二醇还原法相较浸渍法和沉积沉淀法可以使催化剂负载的贵金属金属态更多,进而其活性更好.
制备方法对 Pt-Ir/CeO2 催化氧化甲苯性能的影响
Effect of preparation methods on catalytic performance of Pt-Ir/CeO2 catalyst for toluene oxidation
-
摘要: 负载型贵金属催化剂由于其优异的催化性能而在挥发性有机污染物催化燃烧中被广泛研究. 其中,催化剂制备方法是影响催化剂性能的重要因素. 本文分别采用浸渍法、沉积沉淀法、乙二醇还原法,将等量贵金属 Pt 和 Ir 负载于CeO2 上,探究不同制备方法对催化剂催化氧化甲苯性能的影响. 活性测试结果显示,相较于浸渍法和沉积沉淀法,乙二醇还原法制备的 Pt-Ir/CeO2 催化剂具有最佳的催化反应活性,当Pt、Ir载量为 0.25% wt时,在 180 ℃ 下就能将甲苯完全氧化. 各项表征分析结果表明,乙二醇还原法制备的 Pt-Ir/CeO2-EG 样品由于其 CeO2 表面上具有更多均匀分散的金属态 Pt0 和 Ir0,具有更强的氧活化能力,因此具有更优异的甲苯催化氧化活性.Abstract: Supported noble metal catalysts have been widely researched due to their superior catalytic performance in oxidizing volatile organic compounds. Generally, the preparation method is a crucial factor influencing the catalytic activity of these catalysts. In this study, the same amount of precious metal Pt and Ir were loaded onto CeO2 by impregnation (Pt-Ir/CeO2-IMP), deposition precipitation (Pt-Ir/CeO2-DP) and ethylene glycol reduction method (Pt-Ir/CeO2-EG), respectively, to explore the influence of different preparation methods on the catalytic performance for toluene oxidation on the Pt-Ir/CeO2 catalysts. Compared with Pt-Ir/CeO2-DP and Pt-Ir/CeO2-IMP catalysts, the as-synthesized Pt-Ir/CeO2-EG catalyst exhibited superior catalytic activity, which completely oxidized toluene at a low temperature of 180℃. Various characterization analysis results demonstrated that the Pt-Ir/CeO2-EG catalyst displays a higher oxygen activation capacity with the uniform dispersion of more metallic Pt0 and Ir0 on the surface of CeO2. This work highlights the importance of selecting the appropriate preparation method for achieving optimal catalytic performance in loaded noble metal catalysts.
-
Key words:
- toluene /
- catalytic combustion /
- preparation method /
- PtIr bimetallic catalysts.
-

-

图 1 不同 Pt-Ir/CeO2 催化剂(a)催化氧化甲苯的燃烧曲线;(b)CO2 产率;(c)Pt-Ir/CeO2-EG 催化剂稳定性实验;(d)不同 Pt-Ir/CeO2 催化剂的反应活化能
Figure 1. (a) Light-off curves of toluene combustion over different Pt-Ir/CeO2 catalysts; (b) CO2 conversion rate (c) Tolerance activity for toluene oxidation over Pt-Ir/CeO2-EG catalysts at 165 ℃;(d) Arrhenius plots of different Pt-Ir/CeO2 catalysts

图 2 不同方法制备样品的 XRD 谱图
Figure 2. XRD patterns of the samples prepared by different methods

图 3 3种催化剂和载体的(a)氮气吸脱附等温线图;(b)孔径分布曲线图
Figure 3. (a) The N2 adsorption-desorption isotherms and (b) pore size distribution curves of three catalysts and carrier

图 4 HAADF-STEM 图:(a)Pt-Ir/CeO2-IMP,(c)Pt-Ir/CeO2-DP,(e)Pt-Ir/CeO2-EG;EDS 元素分布图:(b)Pt-Ir/CeO2-IMP,(d)Pt-Ir/CeO2-DP,(f)Pt-Ir/CeO2-EG
Figure 4. HAADF-STEM images and corresponding EDS elemental mapping of (a) and (b) Pt-Ir/CeO2-IMP, (c) and (d) Pt-Ir/CeO2-DP, (e) and (f) Pt-Ir/CeO2-EG

图 5 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 样品 (a) Ir 4f;(b) Pt 4f XPS 图谱
Figure 5. (a) Ir 4f (b) Pt 4f XPS spectra of Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP and Pt-Ir/CeO2-EG
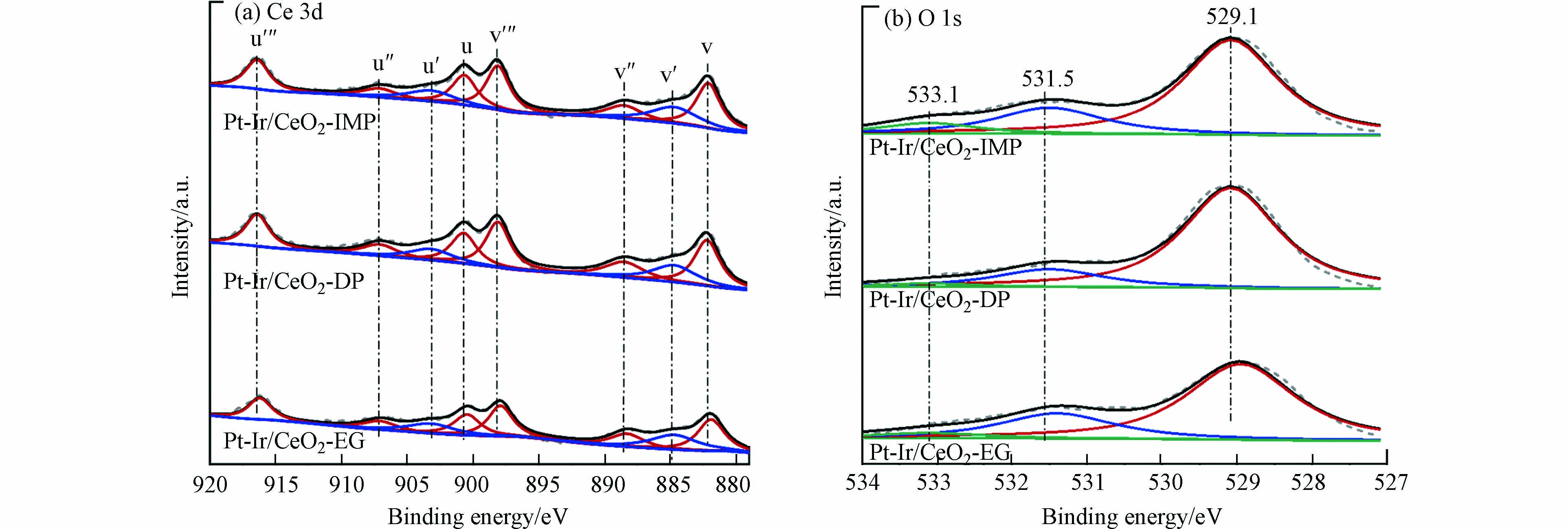
图 6 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 样品 (a) Ce 3d;(b) O 1s XPS 图
Figure 6. (a) Ce 3d (b) O 1s XPS spectra of Pt-Ir/CeO2-IMP, Pt-Ir/CeO2-DP and Pt-Ir/CeO2-EG

图 7 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 样品的 CO-DRIFTS 图
Figure 7. CO-DRIFTS of Pt-Ir/CeO2-IMP, Pt-Ir/CeO2-DP and Pt-Ir/CeO2-EG

图 8 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 样品的(a) O2-TPD 图;(b)H2-TPR 图
Figure 8. (a)O2-TPD and (b)H2-TPR spectra of Pt-Ir/CeO2-IMP, Pt-Ir/CeO2-DP and Pt-Ir/CeO2-EG

图 9 甲苯原位红外图:(a)、(b)Pt-Ir/CeO2-IMP;(c)、(d)Pt-Ir/CeO2-DP;(e)、(f)Pt-Ir/CeO2-EG
Figure 9. In-situ DRIFTS spectra of
1000 ppm toluene oxidation over (a)and(b)Pt-Ir/CeO2-IMP; (c)and(d)Pt-Ir/CeO2-DP and (e)and(f)Pt-Ir/CeO2-EG表 1 3种 Pt-Ir/CeO2 样品中贵金属含量
Table 1. Precious metal content in three Pt-Ir/CeO2 samples
样品
CatalystsPt 含量/%wt
Pt contentIr 含量/%wt
Ir contentPt/Ir 物质的量比 (mol/mol)
Pt/Ir molar ratioPt-Ir/CeO2-IMP 0.30 0.29 1.0 Pt-Ir/CeO2-DP 0.29 0.26 1.1 Pt-Ir/CeO2-EG 0.25 0.20 1.2  下载: 导出CSV
下载: 导出CSV
表 2 3种 Pt-Ir/CeO2 催化剂催化氧化甲苯的特征温度和活化能
Table 2. Characteristic temperature and activation energy of catalytic oxidation of toluene over three Pt-Ir/CeO2 catalysts
样品
CatalystsT10/℃ T50/℃ T90/℃ Ea/(kJ·mol−1) Pt-Ir/CeO2-IMP 136 200 237 75.85 Pt-Ir/CeO2-DP 122 180 223 71.75 Pt-Ir/CeO2-EG 122 148 163 35.88
下载: 导出CSV
表 3 催化剂和载体的 BET 结果
Table 3. BET results for catalysts and support
催化剂
Catalysts比表面积/(m2·g−1)
Surface area孔容/(cm3·g−1)
Pore volume平均孔径/nm
Average pore diameterPt-Ir/CeO2-IMP 64.7 0.22 15.5 Pt-Ir/CeO2-DP 63.6 0.22 15.7 Pt-Ir/CeO2-EG 59.4 0.21 15.4 CeO2 65.1 0.22 15.7
下载: 导出CSV
表 4 Pt-Ir/CeO2-IMP、Pt-Ir/CeO2-DP 和 Pt-Ir/CeO2-EG 样品 XPS 数据
Table 4. The XPS data of Pt-Ir/CeO2-IMP, Pt-Ir/CeO2-DP and Pt-Ir/CeO2-EG
催化剂
Catalysts[Pt0 /(Pt0+ Ptδ+)] /% [Ptδ+ /(Pt0+Ptδ+)] /% [Ir0/(Ir0+Irδ+)]/% [Ce3+/(Ce3++Ce4+)] /% [Oads/(Oads+Olatt)] /% Pt-Ir/CeO2-IMP 14.9 57.8 37.8 22.7 21.6 Pt-Ir/CeO2-DP 33.9 40.9 51.2 20.4 16.3 Pt-Ir/CeO2-EG 72.2 22.2 56.5 25.4 24.7
下载: 导出CSV
-
[1] 刘艺, 宋浩, 刘少俊, 等. 2种制备方法对Pt/Co-Ce催化氧化甲苯性能的影响[J]. 环境工程学报, 2022, 16(5): 1579-1588. LIU Y, SONG H, LIU S J, et al. Effects of two preparation methods on the performance of Pt/Co-Ce catalytic oxidation of toluene[J]. Chinese Journal of Environmental Engineering, 2022, 16(5): 1579-1588 (in Chinese).
[2] LI S J, LIN Y, WANG D, et al. Polyhedral cobalt oxide supported Pt nanoparticles with enhanced performance for toluene catalytic oxidation[J]. Chemosphere, 2021, 263: 127870. doi: 10.1016/j.chemosphere.2020.127870 [3] PENG R S, LI S J, SUN X B, et al. Size effect of Pt nanoparticles on the catalytic oxidation of toluene over Pt/CeO2 catalysts[J]. Applied Catalysis B:Environmental, 2018, 220: 462-470. doi: 10.1016/j.apcatb.2017.07.048 [4] MATSUO K, NUNOTANI N, IMANAKA N. Effect of oxide-ion conductivity of apatite-type Ln10Si6O27 on catalytic activity for toluene combustion[J]. Journal of Asian Ceramic Societies, 2021, 9(4): 1466-1472. doi: 10.1080/21870764.2021.1992850 [5] 黄海凤, 徐琴琪, 陈晓, 等. 整体式Mn基复合金属氧化物催化燃烧VOCs性能研究[J]. 环境化学, 2018, 37(7): 1583-1590. doi: 10.7524/j.issn.0254-6108.2017101307 HUANG H F, XU Q Q, CHEN X, et al. Catalytic combustion of VOCs by integral Mn-based mixed metal oxide[J]. Environmental Chemistry, 2018, 37(7): 1583-1590 (in Chinese). doi: 10.7524/j.issn.0254-6108.2017101307
[6] MO S P, PENG P, PEI Y C, et al. Immobilizing ultrafine bimetallic PtAg alloy onto uniform MnO2 microsphere as a highly active catalyst for CO oxidation[J]. Chinese Chemical Letters, 2021, 32(6): 2057-2060. doi: 10.1016/j.cclet.2020.11.062 [7] ZHANG Y, WU C, WANG Z Q, et al. Enhanced low-temperature catalytic performance for toluene combustion of CeO2-supported Pt-Ir alloy catalysts[J]. Applied Surface Science, 2022, 580: 152278. doi: 10.1016/j.apsusc.2021.152278 [8] ZENG Y Q, WANG Y N, MENG Y H, et al. The effect of preparation method on oxygen activation over Pt/TiO2 catalysts for toluene total oxidation[J]. Chemical Physics Letters, 2019, 730: 95-99. doi: 10.1016/j.cplett.2019.05.048 [9] SUN X C, LIN J, WANG Y H, et al. Catalytically active Ir0 species supported on Al2O3 for complete oxidation of formaldehyde at ambient temperature[J]. Applied Catalysis B:Environmental, 2020, 268: 118741. doi: 10.1016/j.apcatb.2020.118741 [10] 胡涛, 黄琼, 孙月吟, 等. 柠檬酸配合法制备CuMnCeOx化剂及常温催化氧化性能研究[J]. 分子催化, 2022, 36(6): 547-560. HU T, HUANG Q, SUN Y Y, et al. Preparation of CuMnCeO x catalysts by complex method with citric acid and catalytic oxidation at ambient temperature[J]. Journal of Molecular Catalysis, 2022, 36(6): 547-560(in Chinese).
[11] WEN X Y, LI W C, YAN J X, et al. Strong metal–support interaction in Pd/CeO2 promotes the catalytic activity of ethyl acetate oxidation[J]. The Journal of Physical Chemistry C, 2022, 126(3): 1450-1461. doi: 10.1021/acs.jpcc.1c10421 [12] LI Y B, CHEN X Y, WANG C Y, et al. Sodium enhances Ir/TiO2 activity for catalytic oxidation of formaldehyde at ambient temperature[J]. ACS Catalysis, 2018, 8(12): 11377-11385. doi: 10.1021/acscatal.8b03026 [13] 席康, 王勇, 谢晶, 等. 不同Pt前体制备Pt/CeO2催化剂对其结构及性能的影响[J]. 化工学报, 2019, 70(11): 4278-4288. XI K, WANG Y, XIE J, et al. Effect of Pt precursor on structure and performance of Pt/CeO2 catalysts[J]. CIESC Journal, 2019, 70(11): 4278-4288 (in Chinese)
[14] ZHU X Q, HE H, LI Y X, et al. CeO2-supported Pt catalysts derived from MOFs by two pyrolysis strategies to improve the oxygen activation ability[J]. Nanomaterials, 2020, 10(5): 983. doi: 10.3390/nano10050983 [15] KIKKAWA S, TERAMURA K, KATO K, et al. Formation of CH4 at the metal-support interface of Pt/Al2O3 during hydrogenation of CO2: Operando XAS-DRIFTS study[J]. ChemCatChem, 2022, 14(10): e202101723. doi: 10.1002/cctc.202101723 [16] CAO W X, LIN L, QI H F, et al. In-situ synthesis of single-atom Ir by utilizing metal-organic frameworks: An acid-resistant catalyst for hydrogenation of levulinic acid to γ-valerolactone[J]. Journal of Catalysis, 2019, 373: 161-172. doi: 10.1016/j.jcat.2019.03.035 [17] WANG Z Q, ZHANG L X, JI J W, et al. Catalytic enhancement of small sizes of CeO2 additives on Ir/Al2O3 for toluene oxidation[J]. Applied Surface Science, 2022, 571: 151200. doi: 10.1016/j.apsusc.2021.151200 [18] CHANG S S, JIA Y, ZENG Y Q, et al. Effect of interaction between different CeO2 plane and platinum nanoparticles on catalytic activity of Pt/CeO2 in toluene oxidation[J]. Journal of Rare Earths, 2022, 40(11): 1743-1750. doi: 10.1016/j.jre.2021.10.009 [19] LI J, XIA T T, XU J C, et al. Boosting the plasma catalytic performance of CeO2/γ-Al2O3 in long-chain alkane VOCs via tuning the crystallite size[J]. Applied Surface Science, 2023, 611: 155742. doi: 10.1016/j.apsusc.2022.155742 [20] TAN W, XIE S H, CAI Y D, et al. Transformation of highly stable Pt single sites on defect engineered ceria into robust Pt clusters for vehicle emission control[J]. Environmental Science & Technology, 2021, 55(18): 12607-12618. [21] ZHANG Q F, ZHOU Z B, FANG T, et al. Understanding the role of tungsten on Pt/CeO2 for vinyl chloride catalytic combustion[J]. Journal of Rare Earths, 2022, 40(9): 1462-1470. doi: 10.1016/j.jre.2021.09.012 [22] YOU W, ZHANG Q T, JIA H Y, et al. Insights into the state of ceria during ethanol steam reforming over Ir/CeO2[J]. Catalysis Science & Technology, 2023, 13(2): 558-572. [23] CHEN B X, WANG B F, SUN Y H, et al. Plasma-assisted surface interactions of Pt/CeO2 catalyst for enhanced toluene catalytic oxidation[J]. Catalysts, 2018, 9(1): 2. doi: 10.3390/catal9010002 [24] MO S P, LI J, LIAO R Q, et al. Unraveling the decisive role of surface CeO2 nanoparticles in the Pt-CeO2/MnO2 hetero-catalysts for boosting toluene oxidation: Synergistic effect of surface decorated and intrinsic O-vacancies[J]. Chemical Engineering Journal, 2021, 418: 129399. doi: 10.1016/j.cej.2021.129399 [25] LU A L, SUN H L, ZHANG N W, et al. Surface partial-charge-tuned enhancement of catalytic activity of platinum nanocatalysts for toluene oxidation[J]. ACS Catalysis, 2019, 9(8): 7431-7442. doi: 10.1021/acscatal.9b01776 [26] WEI X Q, LI K, ZHANG X Y, et al. CeO2 nanosheets with anion-induced oxygen vacancies for promoting photocatalytic toluene mineralization: Toluene adsorption and reactive oxygen species[J]. Applied Catalysis B:Environmental, 2022, 317: 121694. doi: 10.1016/j.apcatb.2022.121694 [27] MO S P, ZHANG Q, LI J Q, et al. Highly efficient mesoporous MnO2 catalysts for the total toluene oxidation: Oxygen-Vacancy defect engineering and involved intermediates using in situ DRIFTS[J]. Applied Catalysis B:Environmental, 2020, 264: 118464. doi: 10.1016/j.apcatb.2019.118464 [28] ZHANG L X, ZHU Z X, TAN W, et al. Thermal-driven optimization of the strong metal–support interaction of a platinum–Manganese oxide octahedral molecular sieve to promote toluene oxidation: Effect of the interface Pt2+–Ov–Mn δ+[J]. ACS Applied Materials & Interfaces, 2022, 14(51): 56790-56800. [29] SUN W J, HUANG Y J, LI X M, et al. Catalytic combustion of toluene over highly dispersed Cu-CeOx derived from Cu-Ce-MOF by EDTA grafting method[J]. Catalysts, 2021, 11(4): 519. doi: 10.3390/catal11040519 [30] LU J H, ZHONG J P, REN Q M, et al. Construction of Cu-Ce interface for boosting toluene oxidation: Study of Cu-Ce interaction and intermediates identified by in situ DRIFTS[J]. Chinese Chemical Letters, 2021, 32(11): 3435-3439. doi: 10.1016/j.cclet.2021.05.029 [31] REN Q M, ZHAO X Y, ZHONG J P, et al. Unravelling the role of oxygen species in toluene oxidation over Co3O4-base catalysts: in situ DRIFTS coupled with quasi in situ XPS[J]. Journal of Catalysis, 2023, 418: 130-140. doi: 10.1016/j.jcat.2023.01.002 [32] LI M Y, ZHANG W L, ZHANG X W, et al. Influences of different surface oxygen species on oxidation of toluene and/or benzene and their reaction pathways over Cu-Mn metal oxides[J]. Journal of Colloid and Interface Science, 2023, 630: 301-316. doi: 10.1016/j.jcis.2022.10.107 [33] WANG Z W, MA P J, ZHENG K, et al. Size effect, mutual inhibition and oxidation mechanism of the catalytic removal of a toluene and acetone mixture over TiO2 nanosheet-supported Pt nanocatalysts[J]. Applied Catalysis B:Environmental, 2020, 274: 118963. doi: 10.1016/j.apcatb.2020.118963 [34] LIANG W J, ZHU Y X, REN S D, et al. Enhanced catalytic elimination of chlorobenzene over Ru/TiO2 modified with SnO2—Synergistic performance of oxidation and acidity[J]. Chemical Physics, 2023, 566: 111787. doi: 10.1016/j.chemphys.2022.111787 [35] YU K, DENG J, SHEN Y J, et al. Efficient catalytic combustion of toluene at low temperature by tailoring surficial Pt0 and interfacial Pt-Al(OH) x species[J]. iScience, 2021, 24(6): 102689. doi: 10.1016/j.isci.2021.102689 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1578
- HTML全文浏览数: 1578
- PDF下载数: 81
- 施引文献: 0


