

-
大气中悬浮的颗粒物粒径分布范围广,可从3 nm以下到100 μm。其中,100 nm以下的颗粒物通常被称为大气超细颗粒物(UFP)。一方面,UFP粒径小、表面积大,可沉降在呼吸系统深处并对人体健康产生较大危害[1];另一方面,UFP数浓度高,对大气云凝结核数浓度影响显著进而影响着全球气候[2]。UFP数浓度通常主导了大气颗粒物的总数浓度。比如,在北京市城区站点[3-4],UFP质量浓度对大气颗粒物总质量浓度的贡献较小,其数浓度对大气颗粒物总数浓度贡献显著(如图1)。因此,虽然目前广泛研究的大气可悬浮颗粒物(TSP)、PM10、PM2.5中均包含UFP,但对于这些颗粒物的基于质量浓度的认识并不能反映UFP的变化特点。大气UFP包括人为源和天然源排放的一次颗粒物和大气中新粒子生成(new particle formation, NPF)产生的二次颗粒物[5-7]。我国大气污染控制措施不断加严,但对颗粒物的控制主要集中在PM2.5以及颗粒物质量浓度上,对UFP及颗粒物数浓度的关注还相对较少。近年来我国已经把颗粒物数浓度排放限值相继纳入到轻型汽车[8]和重型柴油车[9]等尾气排放标准,国际民航组织等也在推进将颗粒物数浓度纳入航空发动机尾气排放标准[10]。尽管这些排放标准有利于大气一次UFP的控制,由NPF产生的二次UFP的控制却有着较大挑战:例如研究表明尽管北京市大气污染防治取得了非常好的进展,2004到2018年以来NPF发生的频率并没有降低[3, 11]。因此,需要增强对大气UFP的研究和认识。
大气UFP对人体健康和气候的影响取决于其粒径分布、化学组分和形貌等特征,而大气UFP具有来源和组分复杂、时空变化剧烈、化学组分测量困难等特点。从气溶胶生成动力学来看,无论是在污染源烟气中还是大气中,UFP都是气态前体物通过成核和生长过程转化成大颗粒物时的必经之地。而在不同的污染源或大气条件下,气态前体物存在很大差别,导致生成的UFP组分也非常复杂。另一方面,UFP的扩散效应强、停留时间短,加上气态前体物的急剧变化,使得UFP的粒径分布和化学组分具有很强的时空变化特点。然而,由于UFP的质量浓度低、扩散效应强、难与大颗粒物分离等特点,其化学组分的测量,尤其是分子水平的在线测量还非常缺乏。
本文总结了污染源排放的一次UFP特征、大气NPF产生的二次UFP特征、以及实际大气中UFP化学组分及其来源解析的相关研究。分析归纳了目前尚存在的问题,并对未来的研究方向进行了展望。
-
化石燃料、生物质燃料等的燃烧是污染源中UFP的主要产生方式。对全球大气环境和人体健康存在普遍影响的一次颗粒物来源包括:机动车排放(汽油、柴油等燃烧)、生物质燃烧、煤炭燃烧、食物烹饪等,图2统计了这些污染源排放的颗粒物数浓度粒径分布的峰值粒径或几何中值粒径[12-32]。其它污染源如天然气燃烧、轮船排放、航空发动机排放、垃圾焚烧、香烟烟雾、烟花爆竹等以及一些天然源如海盐气溶胶、生物代谢等也会排放一定量的UFP。
机动车排放对城市大气UFP数浓度的影响显著。台架实验与跟车实验结果表明,机动车排放的颗粒物常呈现双模态分布[12-17](图2)。较大模态大多在30—100 nm之间,是机动车内燃机中燃烧产生的颗粒,与其它燃烧源产生的颗粒物粒径相似;较小的模态低至10 nm以下,其形成可能与机动车汽油/柴油燃烧产生的气态硫氧化物、碳氢化合物的成核与冷凝生长有关[33-34]。
生物质燃烧包含森林野火、农业废料燃烧、民用生物燃料燃烧等,是全球最大的有机气溶胶排放源之一[35]。生物质燃烧大部分属于无组织燃烧,燃烧条件不受控且没有污染物控制措施,具有明显的季节特征,如用于取暖的生物质燃烧多发生在冬季、农业废料的燃烧则集中在农产品收获季节之后、森林野火多发生在干燥的季节等。生物质燃烧排放的颗粒物峰值粒径大多在30—90 nm之间[18-24]。
煤炭是许多国家的主要能源类型,煤炭燃烧主要包括电厂、工业锅炉、集中供暖等用途的工业煤燃烧和炊事、取暖等用途的民用煤燃烧。工业煤燃烧一般具有燃烧条件稳定、燃烧温度高、氧气充足、安装有除尘措施等特点,而民用煤燃烧常存在燃烧温度低、燃烧不充分、无污染物控制措施等情况[36]。二者(特别是民用煤燃烧)对大气颗粒物一次排放均有较大贡献。煤炭燃烧排放的颗粒物峰值粒径大多在30—70 nm之间[23-27]。
食物烹饪包括居民厨房、商业餐厅、露天烧烤等,对室内和室外空气污染均有较大贡献。不同于固体燃料燃烧,烹饪排放的颗粒物通常指食用油、动植物油脂等在烹饪过程中产生的颗粒物,而不包含生火过程燃料燃烧产生的颗粒物。烹饪排放具有显著的日变化特征,通常在每日的早、中、晚三餐时间出现峰值。食物烹饪排放的颗粒物峰值粒径大多在30—100 nm之间[23-27]。
综上所述,燃烧过程和高温过程大多会产生峰值粒径在30—100 nm范围内的颗粒物模态,近年来逐渐引起较多关注的航空发动机[10, 37]和轮船[38-39]排放的颗粒物粒径峰值大多也在该范围内。而机动车排放还观测到一个峰值粒径低至10 nm以下的模态,在大气颗粒物源解析中常被用作机动车排放源的判定[40-41]。不同研究中颗粒物粒径分布的差异来源于燃料种类、燃烧方式、排放控制技术等的不同。比如,机动车的发动机类型、车辆运行工况和稀释条件不同,颗粒物的排放强度和粒径分布也不同[33, 42-43];民用燃煤或生物质燃烧中,提高燃烧炉的能量效率、使用新型炉具和清洁燃料、原煤制成煤砖/半焦等措施可大大减少颗粒物排放、改变颗粒物粒径分布[19, 44-47];在食物烹饪中,煎炸、快炒的烹饪方式通常会产生较多的UFP,而蒸煮产生的UFP则很少[48-49]。
-
燃烧过程中产生的颗粒物主要由有机物、黑炭(元素碳)、无机盐、金属氧化物等构成。燃料中可燃有机组分的不完全燃烧会产生黑炭颗粒和大量有机气态前体物,非可燃的无机组分则在高温下气化生成无机气态前体物,有机和无机的气态前体物可均相成核形成颗粒物,或在已生成的颗粒物表面冷凝。由此产生的无机颗粒、黑炭颗粒、有机颗粒和多种成分混合的颗粒物又会发生聚并和进一步冷凝生长,从而使颗粒物粒径变大,组分更加复杂[50-51]。
针对污染源排放UFP化学组分的研究非常缺乏,大多数研究都是基于污染源产生的总颗粒物组分或PM2.5组分。不同燃料的燃烧过程或高温气化过程会产生不同的气态前体物和颗粒物组分,UFP和PM2.5或其它粒径颗粒物组分在很大程度上都取决于气态前体物类别,因此PM2.5等颗粒物组分可为UFP组分研究提供一定的参考。表1汇总了不同排放源产生的颗粒物组分。首先,化石燃料的不完全燃烧都会产生烷烃、烯烃、黑炭和多环芳烃等物质,其中黑炭和多环芳烃可作为不完全燃烧过程的示踪物[52]。机动车通常采用汽油和柴油作为燃料,排放的气态和颗粒态产物中含有较多的芳香烃类物质和含氮组分,如硝酸盐、有机胺、硝基多环芳烃等;藿烷、甾烷、晕苯、异喹啉等常被作为机动车燃烧的示踪物[52-56]。柴油车排放的黑炭多于其它燃料的机动车和其它燃烧源,通常是大气中黑炭的主要来源。煤炭燃烧的示踪物较少,有研究表明,Si, Se, As, Cr, Co, Cu, Al等氧化物在煤炭燃烧中的排放大于其它排放源[52]。生物质燃料的种类多种多样:含有较多纤维素、木质素等的生物质燃烧会产生左旋葡聚糖、甲氧基苯酚、愈创木酚等[52];K+是维持植物细胞平衡的重要离子,因此钾元素也是生物质燃烧的示踪物。油酸、亚油酸、棕榈酸等饱和或不饱和脂肪酸常被用作烹饪的示踪物,胆固醇是肉类烹饪的示踪物[48]。
综上所述,各种污染源排放的颗粒物成分复杂、包含一些相似组分和各种特征物质/示踪物,后者常被用在大气颗粒物源解析中辅助识别一次来源。然而,污染源排放的UFP组分是否与100 nm以上的颗粒物组分存在较大差别有待研究,这依赖于对前述复杂组分的分子水平的高物种分辨率测量和识别。
-
污染源产生颗粒物的粒径分布和化学组分会随时间会发生剧烈变化。以煤燃烧为例,Zhou等[25]研究了神木烟煤燃烧产生的颗粒物粒径分布和化学组分随时间的变化(如图3):燃烧过程可分为点火、剧烈燃烧、稳定燃烧和燃尽四个阶段。前两阶段燃煤中的有机挥发分快速脱出,产生的颗粒物粒径较大,主要贡献了总排放颗粒物的质量浓度,颗粒物中有机组分为主导;而稳定燃烧阶段燃烧效率最高,产生颗粒物粒径较小,主要贡献了总排放颗粒物的数浓度,颗粒物中无机组分占比增加而有机气组分的氧化程度不断升高。
污染源颗粒物排放到大气后还会经过一系列复杂的大气迁移转化过程,使其粒径分布和化学组分在短时间内发生剧烈变化。首先,UFP的扩散效应强、大气停留时间短,排放到大气后极易与大气中的背景气溶胶发生凝并作用,使粒径分布发生较大改变;其次,污染源排放的UFP和气态污染物在与大气混合后,会经历气固分配过程,使颗粒物组分发生改变;最后,一次排放的UFP中有较多未完全燃烧的氧化程度较低的成分,在大气中会被O3、羟基自由基、硝基自由基等氧化,该过程又称为大气颗粒物的老化。例如,离交通源越远的大气站点往往其UFP数浓度越低[58]、粒径越大[59]、二次颗粒物比例越高[60]。
综上所述,污染源排放的UFP粒径分布和化学组分不仅在产生过程中会在数分钟至数小时内发生剧烈变化,其在排放到大气后也会经过复杂的变化。因此,有必要对其进行小时量级或分钟量级上的高时间分辨率变化研究。
-
大气NPF是指大气中的气态前体物通过成核和生长产生二次颗粒物的现象。大气NPF可在数小时内产生大量二次UFP,该过程贡献了全球大气颗粒物总数浓度的一半以上[2]。
大气二次UFP的化学组分主要取决于参与成核和生长的大气气态前体物及其反应产物。硫酸(H2SO4)被广泛认为是大气中成核的关键气态前体物。主要来自于大气中SO2的光化学氧化反应,具有较低的饱和蒸气压,可以与大气中H2O、氨气、有机胺、有机物等形成氢键或离子键而成核[61-64]。硫酸与氨气或有机胺成核的过程通常被称为酸碱成核,被认为是城市大气环境里的重要成核过程[3, 63, 65-66]。除硫酸以外,甲基磺酸(MSA)、有机羧酸等也可以作为酸性气体参与酸碱成核[61, 67]。近期有文章通过烟雾箱实验和模型模拟提出,在温度较低的平流层或城市地区低温条件下,挥发性较高的硝酸也可以作为酸性气体参与成核和二次粒子的快速生长[68]。
大气中有机物也可以发生均相成核和生长。有机气态前体物贡献成核和生长的能力主要取决于其挥发性[69]。一般来说,挥发性越低的有机物越容易贡献均相成核和冷凝生长。基于有机物的饱和蒸气压(C*),可以把有机气体分为超低挥发性的有机物(ELVOC, C* < 3×10−4 μg·m−3)、低挥发性有机物(LVOC, 3×10−4 < C* < 0.3 μg·m−3)、半挥发性有机物(SVOC, 0.3 < C* < 300 μg·m−3)、中间挥发性有机物(IVOC, 300 < C* < 3×106 μg·m−3)以及挥发性有机物(VOC, C* > 3×106 μg·m−3)[69-70]。其中,ELVOCs通常含氧量较高(O:C > 0.8),是贡献纳米颗粒物初期生长的重要前体物。然而,大气中纯有机物成核很少被观测到,往往不能排除大气中普遍存在的硫酸引起的酸碱成核对NPF的贡献。
除了中性的酸性、碱性气体和有机气体可以参与新粒子生成和生长以外,有机和无机大气离子也可以通过静电作用促进大气分子结合形成团簇而参与到离子诱导成核中[71-72],这种现象主要发生在中性气态前体物浓度低而离子浓度比较高时,比如高海拔地区[73]或对流层顶[74]。
-
大气NPF产生的新粒子也具有很强的时间变化特征。大气NPF通常发生在一天中光照最强时,此时光化学反应产生大量新粒子生成和生长所需的气态前体物。此时,在粒径谱上可以观察到3 nm以下颗粒物的爆发式出现,随后这些新生成的颗粒物通过冷凝生长和聚并过程使得峰值粒径不断增大,当峰值粒径增长到20—100 nm以后,数浓度粒径分布不再有显著变化,这个过程持续数小时至数天。大气NPF在全球范围内普遍发生并产生大量的UFP,3 nm颗粒物的生成速率在0.01—100 #·cm−3·s−1[75],新粒子生长速率在1—20 nm·h−1[75]。而城市大气环境里1.5 nm颗粒物的生成速率可超过1000 #·cm−3·s−1[76-77]。相对于污染源一次UFP的显著空间变化特点,大气NPF常常具有显著的区域性特点,即在较大范围内的稳定大气环境中均可发生NPF,主要取决于气态前体物浓度、背景气溶胶浓度、温湿度等因素。如北京的研究结果表明低气溶胶背景浓度[78]和低温[3]有利于NPF的发生。
新粒子生成和生长不同阶段二次UFP的组分可能会发生较大变化。在H2SO4-NH3、H2SO4-二甲胺、MSA-NH3-有机胺成核的实验中[66-68],发现10—15 nm以下的颗粒物酸碱比例大于1而颗粒物逐渐增长到10—15 nm以上时酸碱比例接近于1;而在HNO3-二甲胺成核的实验中[69],颗粒物在9—30 nm的粒径范围内一直呈现酸碱比为1∶1的状态。不同酸碱成核体系下颗粒物不同粒径酸碱比的差异可能与酸性气体的酸性、挥发性和酸碱团簇的稳定性等因素有关。Winkler等[79]测量了天然源挥发性有机物(BVOCs)在烟雾箱中氧化产生的10—40 nm颗粒态的化学组分,发现较小粒径中含有大量有机酸,而较大粒径中含有较多羰基化合物和小分子有机酸。实际大气中气态前体物种类复杂,可能存在多种成核和生长机制。Ehn等[80]根据酸碱成核和有机物冷凝生长推测了大气新粒子生长过程化学组分的变化(如图4)。结果表明,硫酸和碱性气体在成核初期发挥主要作用,而ELVOCs、LVOCs和SVOCs等在新粒子生长过程依次发挥重要作用。
综上所述,基于实验室的成核实验和理论模拟,已经认识到硫酸等酸性气体、氨和有机胺等碱性气体、超低挥发性和低挥发性有机物等是大气新粒子生成的重要气态前体物。然而,由于UFP化学组分在线测量仪器的缺乏,实际大气中对于新粒子生成和生长过程化学组分变化的直接观测结果还非常缺乏.
-
综上所述,无论是污染源排放还是新粒子生成过程产生的UFP,都具有组分复杂和随时间剧烈变化的特征。对大气UFP组分的在线测量是认识其来源构成和大气迁移转化规律的基础。然而,UFP组分测量的两个难点大大制约了其测量技术的发展:(1)UFP扩散效应强,与大颗粒、气态组分的高效分离困难;(2)UFP质量小,需要高灵敏度测量技术。目前化学组分的测量还是以离线分析为主,通过多级惯性撞击器(MOUDI)最后一级[81]、纳米多级惯性撞击器(Nano MOUDI)最后2-3级、静电低压惯性撞击器(ELPI)、大流量撞击器[82]等将UFP与大颗粒物分离并捕集在滤膜上,萃取后使用离子色谱、OC/EC分析仪等进行测试。UFP化学组分的在线测量目前没有商业化的仪器,Smith等[83]、Wagner等[84]和陈等[85]详细总结了UFP组分在线测量仪器及其优缺点。Smith等[86-87]研发的静电捕集热脱附化学电离质谱(TDCIMS)可测量不同粒径范围内的UFP化学组分,测量原理相似的仪器还有热脱附-离子漂移-化学电离质谱(TD-ID-CIMS)[88],热脱附-差分电迁移率-化学电离质谱(TD-DMA)[84],带电颗粒物化学分析仪(CAChUP)[89]等。Li等[4]通过使用软X射线代替放射性元素作为电离源改进了TDCIMS使其可用于放射性物质严格管制的地区,同时对TDCIMS测量各粒径UFP的效率和对各物质的灵敏度进行了较全面的标定,并在北京开展观测实现了大气UFP的分子水平在线测量。Wang和Johnston等[90]研发的纳米气溶胶质谱(NAMS)可用来测量UFP单颗粒的元素组成。另外,气体和颗粒物滤膜采集-化学电离质谱(FIGAERO-CIMS)[91]和萃取电喷雾质谱(EESI-MS)[92-93]等商业化仪器虽然可以测量有机颗粒物的分子水平化学组分,却因为缺乏大颗粒物的分离单元而无法直接用于大气UFP的测量。
-
目前大气UFP化学组分的定量分析大多基于离线测量,图5总结了不同地区使用MOUDI测量的大气中56—100 nm的UFP化学组分。这些站点的UFP质量浓度在0.6—7.71 μg·m−3范围内。其中,有机组分在所识别的总组分中占20%—70%,在大多数站点为主要成分。其它组分还包含黑炭2%—18%,硫酸盐0—20%,硝酸盐1%—26%,铵盐0—33%,金属离子或金属氧化物0—27%。北京UFP中有机组分比例高于100 nm以上亚微米颗粒物有机组分的比例[94-98],这可能解释了北京UFP的吸湿因子低于100 nm以上亚微米颗粒物[97]。在加州圣华谷的多个站点[99-100]也观测到UFP中有机组分高于其它亚微米颗粒物这一现象;而另一些地区,如上海[101]、台湾[102]、加州河边[103]、匹兹堡[104]等地,则没有明显地观测到这一现象。
大气UFP化学组分的在线测量主要使用质谱方法。Smith[105]等使用TDCIMS首次实现了大气中20 nm以下颗粒物的在线观测,发现美国亚特兰大[105]和科罗拉多博尔德[86]在NPF期间大气6—20 nm颗粒物化学组分以硫酸铵为主;而墨西哥[106]的NPF期间大气10—33 nm颗粒物中有机组分的信号占主导,北欧森林[107]NPF期间大气20—70 nm颗粒物中主要以单萜烯氧化的有机产物信号为主,且伴随有机羧酸比例增加,北京大气UFP中有机组分占主导并且含氮和含硫的有机组分比例高于其它地区[4];爱尔兰大西洋[108]站点NPF期间大气15—85 nm颗粒物中主要以有机组分和NaCl的信号为主,特拉华州刘易斯市[109]TDCIMS和NAMS的联合观测表明低挥发性有机组分贡献了一半以上新粒子生长,且包括了较多含氮有机组分。尽管如此,NAMS在该地区的结果显示氮、硫元素比例在NPF期间会升高,显示了硫酸铵和硝酸铵对新粒子生长的贡献。Smith等[110]和Lawle等[107]通过TDCIMS测量发现有机胺盐在多个城市、乡村、森林站点的大气纳米颗粒物中普遍存在。通过TDCIMS在美国南部平原站点[111]的观测表明了新粒子生长的三类途径,即有机物贡献生长,硫酸铵贡献生长,硫酸、碱基和有机物共同贡献生长。综合以上观测结果,硫酸铵、有机胺盐、有机组分在新粒子生长过程中发挥了重要作用。然而,NAMS的测量只能提供元素组成,而以上TDCIMS的结果大多基于质谱原始信号强度,缺乏准确的定量结果。
-
目前对于大气UFP的来源解析研究主要基于离线UFP化学组分测量、在线UFP化学组分测量和UFP粒径分布测量。
基于离线大气UFP化学组分测量的源解析工作多集中在美国加州等少数地区,且主要针对源排放一次UFP。在加州境内的多个站点[112-114]分别使用MOUDI将56—100 nm的UFP采集在离线滤膜上,时间分辨率为数小时至数天。分别使用左旋葡聚糖、胆固醇、藿烷/甾烷、EC作为生物质燃烧、肉类烹饪、汽油车、柴油车排放的示踪物,结合化学质量平衡(CMB)受体模型进行源解析,得到图6所示不同一次源对于UFP有机碳的贡献或对UFP有机碳和元素碳总和的贡献。可看出肉类烹饪、汽油车、柴油车和木柴燃烧等是美国加州各个地区大气UFP或其中有机组分的主要来源,分别占0—67%(均值23%)、0—25%(均值12%)、0—21%(均值8%)、0—79%(均值25%)。由于尚未能确定NPF生成的二次UFP的有效源谱或示踪物,因此基于CMB的分析无法识别NPF对大气UFP的贡献。图6中其它未知来源可能包含了NPF的贡献。总体来看,未知来源贡献日间比夜间高[113]、夏季比冬季高[112],可能同日间和夏季光照更强、更有利于二次UFP生成有关。
基于在线UFP化学组分测量的来源解析工作非常少,且缺乏准确定量的结果。Glicker等[115]使用TDCIMS测量亚马逊地区大气UFP化学组分,结合后向轨迹发现当站点受到人为源影响时,硫酸盐和氯离子的信号比例会升高;而在不受人为源影响时,信号强度最高的物质为异氰酸根、醋酸根、草酸根以及异戊二烯氧化特征产物的热解碎片C5H7O+。Lawler等[116]在美国俄克拉荷马州北部的农业区检测到大量甲壳素的热解产物和C6糖醇,这些物质可能对应真菌孢子破碎释放的粒径峰值约30 nm的UFP。然而,由于缺乏全面的标定,尚无法准确定量不同源对于UFP的贡献,并且不同研究之间的可比性较差。
在缺少大气UFP化学组分测量的情况下,一些研究基于大气UFP的数浓度粒径分布对其进行来源解析,并结合气体浓度或亚微米颗粒物组分判别来源类型。如Liu等[41]和Wang等[40]对颗粒物粒径分布进行正交矩阵因子分析(PMF)得出大气颗粒物由多个模态组成,结合NO2浓度、黑炭浓度、PM1中解析出的食物烹饪源、生物质燃烧源、二次气溶胶源等时间序列,得到大气UFP主要由机动车源、食物烹饪源、燃烧源、二次气溶胶等组成。Brines等[117]通过对大气UFP粒径分布进行聚类分析发现交通源、二次生成源、以及背景气溶胶主导大气UFP的时间分别占44%—63%、14%—19%和7%—22%。Kontkanen等[118]利用气溶胶通用动力学方程对粒径谱上突然出现的10—30 nm颗粒物进行定量分析,并通过对比NOx变化推测出其来自于机动车排放。虽然基于粒径谱的源解析分析可以得出各个粒径分布模态对于大气UFP的贡献比例,然而从图2可知,不同源产生的颗粒物粒径分布重合较多,因此在复杂大气环境下仅从粒径分布上解析不同源的贡献是需要谨慎对待的。
-
目前研究者们已经在大气UFP一次来源和二次来源特征及化学组分上取得了一定的研究进展,然而对大气UFP化学组分在分钟和小时量级上的高时间分辨率变化、分子水平上的高物种分辨率的认识还十分有限。目前对各种来源的UFP组分是否与大粒径颗粒物存在显著差别、大气中UFP的来源贡献、UFP在大气中发生演化的关键气态前体物和影响因素等问题认识不清。未来需要从以下方面加强对UFP化学组分、来源及其大气迁移转化过程的认识,为UFP健康效应和气候效应的评估和相关控制措施的制定提供基础数据和科学支撑。
(1)加强对大气UFP多组分分子水平的研究。对UFP来源及其健康效应和气候效应的研究需要获取分子水平的有机组分信息。然而,目前对于UFP有机组分的分析大多停留在总有机碳、分子碎片、或少数示踪物的测量上,尚未能获取对化学组分多维度的认识,这大大增加了来源解析的不确定性。针对此,应推进分子水平、高物种分辨率的化学组分分析和源解析工作。
(2)加强对大气UFP大气迁移转化过程的认识。一次和二次来源的UFP均有显著的时间变化特征,而目前对于UFP化学组分贡献的定量认识多来源于离线测量,时间分辨率为一天至数天;有限的在线研究在定量、可比性上有待加强。针对此,建议推进在线UFP化学组分测量技术的研发,统一测量方法,开展高时间分辨率的UFP大气迁移转化过程研究。
大气超细颗粒物来源及其化学组分研究进展
Research progress on the sources and the chemical composition of ambient ultrafine particles
-
摘要: 大气超细颗粒物(ultrafine particle, UFP)为粒径小于100 nm的颗粒物,其数浓度主导了大气颗粒物的总数浓度,对人体健康和气候都有显著影响。大气UFP主要来源于污染源烟气和大气中气态前体物的成核和生长。其中,污染源烟气中成核和生长产生的颗粒物排放后构成大气UFP的一次来源,而直接在大气中成核和生长(又称新粒子生成)产生的颗粒物构成大气UFP的二次来源。由于不同来源气态前体物种类的显著差别,所生成UFP的化学组分及其健康和气候效应也会有显著不同。然而,目前对大气UFP的不同来源贡献及其化学组成的相关研究相对较少。本文简要介绍了几种主要污染源排放的一次UFP特征、大气新粒子生成产生的二次UFP特征、以及受多种来源影响的实际大气UFP的化学组分及其来源解析相关研究进展。基于上述分析,建议未来着重开展高时间分辨率和高物种分辨率的大气UFP化学组分观测,加强对大气UFP分子水平上组分及其变化规律的认识。Abstract: The ambient ultrafine particles (UFP) are particles with diameter smaller than 100 nm. Their number concentrations dominate the total ambient particle number concentrations, and have significant effect on human health and the global climate. Ambient UFP are originated from the nucleation and the subsequent growth from the gas precursors either in the plume of the emission sources or in the atmosphere. The UFP formed in the emission plume are referred to as primary UFP. The UFP formed in the atmosphere are referred to as secondary UFP and the whole nucleation and growth process in the atmosphere is called new particle formation (NPF). Due to the different precursors in different emission plumes and in the atmosphere, the chemical composition of the generated UFP and their health and climate effects are very different. However, the source contributions and chemical composition of the ambient UFP are poorly understood. This study briefly summarized the studies on the characteristics of the primary UFP emitted from pollution sources and the secondary UFP generated from NPF events. We also introduced the latest researches on the chemical composition and source apportionment of ambient UFP which involve both primary and secondary UFP. Based on these research progresses, we suggest that ambient UFP measurement with high species-resolution and high time-resolution should be conducted to further understand the molecular level chemical composition and the evolution of ambient UFP.
-

-
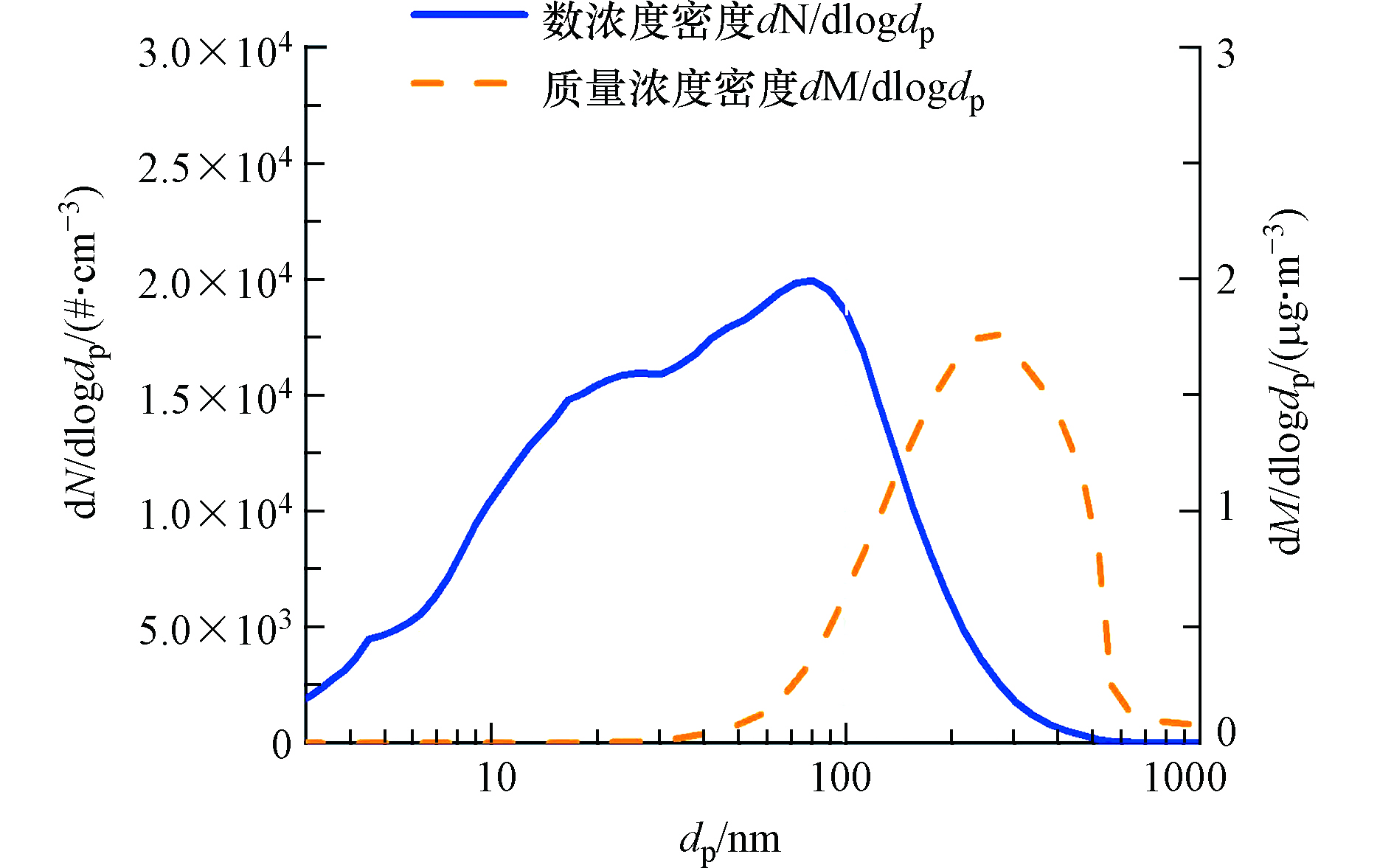


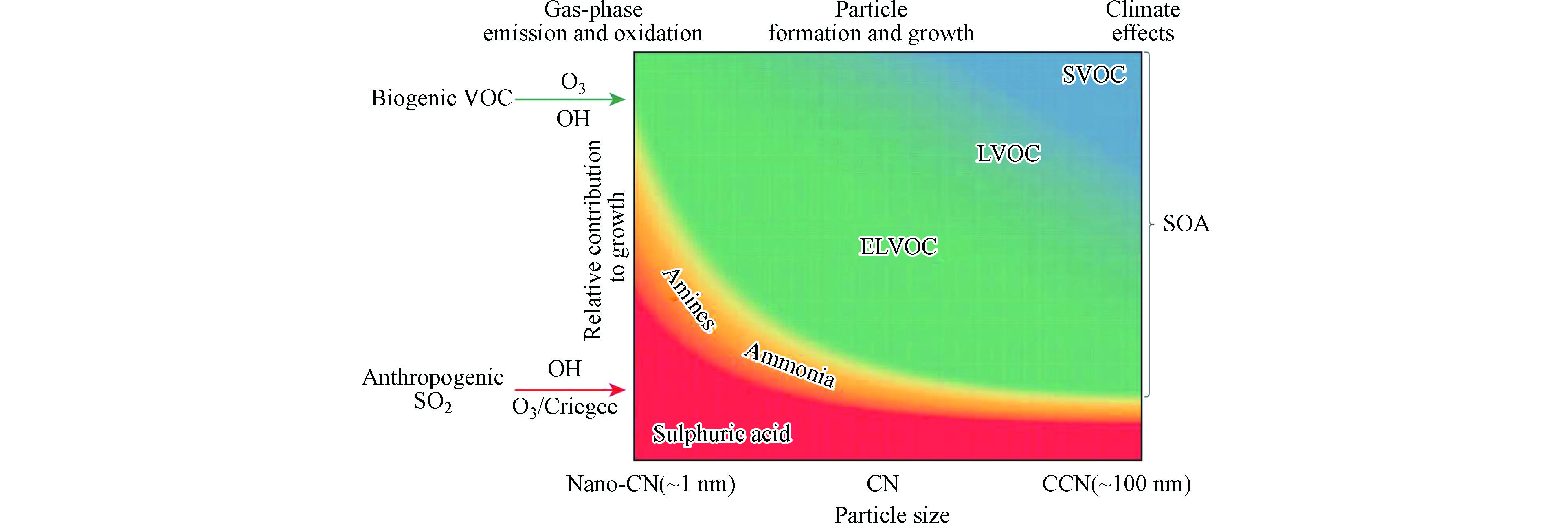
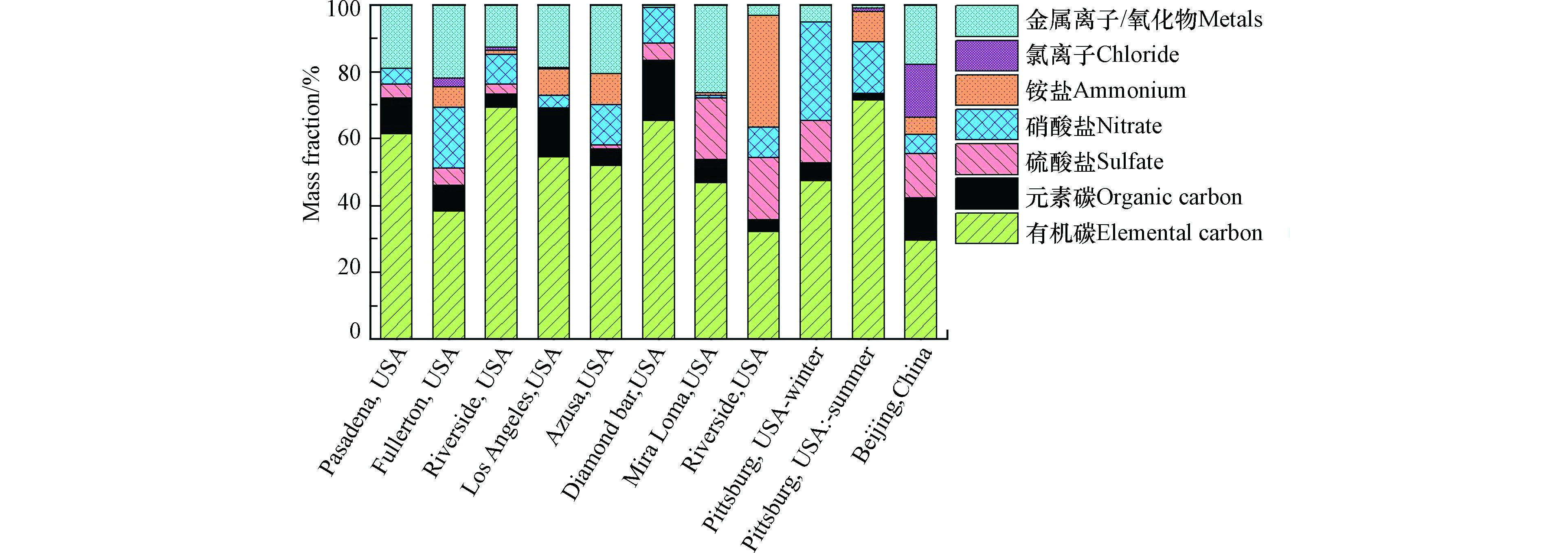
图 5 美国加州[103]、美国匹兹堡[104]、中国北京[94]等站点基于MOUDI与离线滤膜结合测量的56—100 nm颗粒物在所识别的总组分中的占比
Figure 5. The chemical composition fractions of ultrafine particles among the identified species in different stations in California[103], Pittsburg[104], and Beijing[94]. The ultrafine particle samples were collected on filters at the last stage of MOUDI.
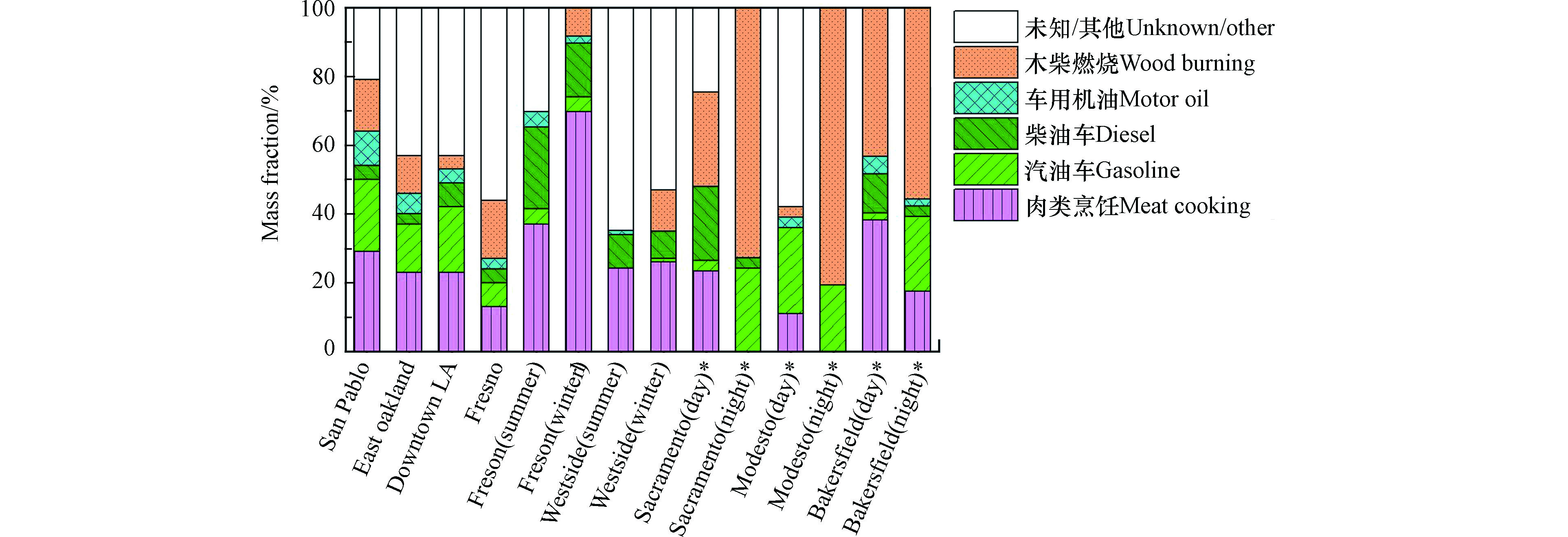
表 1 不同污染源产生颗粒物中主要化学组分及其示踪物
Table 1. The main chemical composition and tracers of particles from different emission sources
排放源
Emission sources主要化学组分
Main chemical composition示踪物/特征物质
Tracers机动车尾气[52-56] 黑炭、芳香烃、多环芳烃、烷烃、烯烃、甾烷、藿烷、硝基多环芳烃、含氧多环芳烃、硝酸盐、铵盐、有机胺盐、金属氧化物等 藿烷、甾烷、晕苯、异喹啉、苯并[ghi]吡、类异戊二烯、三环萜烷、Sb、Br、Zn、Ba、Pb、EC等 生物质燃烧[52, 54, 57] 黑炭、烷烃、烯烃、醛酮类、脂肪酸、脂肪醇、甲氧基苯酚、单糖类及其衍生物、固醇、双萜类、三萜类、蜡酯、多环芳烃、硫酸盐、硝酸盐、铵盐、有机铵盐、氯离子、钠离子、钾离子等 左旋葡聚糖、甲氧基苯酚、蜡酯、K、脱氢松香酸、脱氢枞酸、海松酸、惹烯等、二帖化合物、植物甾醇、愈创木酚等 煤炭燃烧[52] 黑炭、烷烃、醛酮类、有机酸、多环芳烃、硫酸盐、铵盐、有机胺盐、氯离子、金属氧化物等 Si, Se, As, Cr, Co, Cu, Al等 食物烹饪[48] 黑炭、饱和脂肪酸、不饱和脂肪酸、二元羧酸、醇酮类、内酯、芳香烃、有机胺、烷烃、固醇、单糖酐、多环芳烃、呋喃酰胺等 油酸、亚油酸、棕榈酸、C4-C8二羧酸、胆固醇等  下载: 导出CSV
下载: 导出CSV
-
[1] VALAVANIDIS A, FIOTAKIS K, VLACHOGIANNI T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms [J]. Journal of Environmental Science and Health, Part C, 2008, 26(4): 339-362. doi: 10.1080/10590500802494538 [2] MERIKANTO J, SPRACKLEN D V, MANN G W, et al. Impact of nucleation on global CCN [J]. Atmospheric Chemistry and Physics, 2009, 9(21): 8601-8616. doi: 10.5194/acp-9-8601-2009 [3] DENG C J, FU Y Y, DADA L, et al. Seasonal characteristics of new particle formation and growth in urban Beijing [J]. Environmental Science & Technology, 2020, 54(14): 8547-8557. [4] LI X X, LI Y Y, LAWLER M J, et al. Composition of ultrafine particles in urban Beijing: Measurement using a thermal desorption chemical ionization mass spectrometer [J]. Environmental Science & Technology, 2021, 55(5): 2859-2868. [5] KUMAR P, MORAWSKA L, BIRMILI W, et al. Ultrafine particles in cities [J]. Environment International, 2014, 66: 1-10. doi: 10.1016/j.envint.2014.01.013 [6] ZHANG R Y, KHALIZOV A, WANG L, et al. Nucleation and growth of nanoparticles in the atmosphere [J]. Chemical Reviews, 2012, 112(3): 1957-2011. doi: 10.1021/cr2001756 [7] BIRMILI W, WIEDENSOHLER A. New particle formation in the continental boundary layer: Meteorological and gas phase parameter influence [J]. Geophysical Research Letters, 2000, 27(20): 3325-3328. doi: 10.1029/1999GL011221 [8] 中华人民共和国环境保护部, 轻型汽车污染物排放限值及测量方法(中国第五阶段). 2013. [9] 中华人民共和国环境保护部, 重型柴油车污染物排放限值及测量方法(中国第六阶段). 2019. [10] ZHANG X L, CHEN X, WANG J. A number-based inventory of size-resolved black carbon particle emissions by global civil aviation [J]. Nature Communications, 2019, 10: 534. doi: 10.1038/s41467-019-08491-9 [11] LI X X, ZHAO B, ZHOU W, et al. Responses of gaseous sulfuric acid and particulate sulfate to reduced SO2 concentration: A perspective from long-term measurements in Beijing [J]. Science of the Total Environment, 2020, 721: 137700. doi: 10.1016/j.scitotenv.2020.137700 [12] LI T Z, CHEN X D, YAN Z X. Comparison of fine particles emissions of light-duty gasoline vehicles from chassis dynamometer tests and on-road measurements [J]. Atmospheric Environment, 2013, 68: 82-91. doi: 10.1016/j.atmosenv.2012.11.031 [13] SHI J P, HARRISON R M. Investigation of ultrafine particle formation during diesel exhaust dilution [J]. Environmental Science & Technology, 1999, 33(21): 3730-3736. [14] CARPENTIERI M, KUMAR P. Ground-fixed and on-board measurements of nanoparticles in the wake of a moving vehicle [J]. Atmospheric Environment, 2011, 45(32): 5837-5852. doi: 10.1016/j.atmosenv.2011.06.079 [15] VOGT R, SCHEER V, CASATI R, et al. On-road measurement of particle emission in the exhaust plume of a diesel passenger car [J]. Environmental Science & Technology, 2003, 37(18): 4070-4076. [16] GIECHASKIEL B, NTZIACHRISTOS L, SAMARAS Z, et al. Formation potential of vehicle exhaust nucleation mode particles on-road and in the laboratory [J]. Atmospheric Environment, 2005, 39(18): 3191-3198. doi: 10.1016/j.atmosenv.2005.02.019 [17] BISWAS S, HU S H, VERMA V, et al. Physical properties of particulate matter (PM) from late model heavy-duty diesel vehicles operating with advanced PM and NOx emission control technologies [J]. Atmospheric Environment, 2008, 42(22): 5622-5634. doi: 10.1016/j.atmosenv.2008.03.007 [18] HOSSEINI S, LI Q, COCKER D, et al. Particle size distributions from laboratory-scale biomass fires using fast response instruments [J]. Atmospheric Chemistry and Physics, 2010, 10(16): 8065-8076. doi: 10.5194/acp-10-8065-2010 [19] TISSARI J, LYYRÄNEN J, HYTÖNEN K, et al. Fine particle and gaseous emissions from normal and smouldering wood combustion in a conventional masonry heater [J]. Atmospheric Environment, 2008, 42(34): 7862-7873. doi: 10.1016/j.atmosenv.2008.07.019 [20] WARDOYO A Y P, MORAWSKA L, RISTOVSKI Z D, et al. Quantification of particle number and mass emission factors from combustion of Queensland trees [J]. Environmental Science & Technology, 2006, 40(18): 5696-5703. [21] WIINIKKA H, GEBART R. Critical parameters for particle emissions in small-scale fixed-bed combustion of wood pellets [J]. Energy & Fuels, 2004, 18(4): 897-907. [22] TIWARI M, SAHU S K, BHANGARE R C, et al. Particle size distributions of ultrafine combustion aerosols generated from household fuels [J]. Atmospheric Pollution Research, 2014, 5(1): 145-150. doi: 10.5094/APR.2014.018 [23] ZHANG H F, WANG S X, HAO J M, et al. Chemical and size characterization of particles emitted from the burning of coal and wood in rural households in Guizhou, China [J]. Atmospheric Environment, 2012, 51: 94-99. doi: 10.1016/j.atmosenv.2012.01.042 [24] ZHANG H F, ZHU T, WANG S X, et al. Indoor emissions of carbonaceous aerosol and other air pollutants from household fuel burning in southwest China [J]. Aerosol and Air Quality Research, 2014, 14(6): 1779-1788. doi: 10.4209/aaqr.2013.10.0305 [25] ZHOU W, JIANG J K, DUAN L, et al. Evolution of submicrometer organic aerosols during a complete residential coal combustion process [J]. Environmental Science & Technology, 2016, 50(14): 7861-7869. [26] GAO Q, LI S Q, YANG M M, et al. Measurement and numerical simulation of ultrafine particle size distribution in the early stage of high-sodium lignite combustion [J]. Proceedings of the Combustion Institute, 2017, 36(2): 2083-2090. doi: 10.1016/j.proci.2016.07.085 [27] MASEKAMENI D M, BROUWER D, MAKONESE T, et al. Size distribution of ultrafine particles generated from residential fixed-bed coal combustion in a typical brazier [J]. Aerosol and Air Quality Research, 2018, 18(10): 2618-2632. doi: 10.4209/aaqr.2018.03.0105 [28] BUONANNO G, MORAWSKA L, STABILE L. Particle emission factors during cooking activities [J]. Atmospheric Environment, 2009, 43(20): 3235-3242. doi: 10.1016/j.atmosenv.2009.03.044 [29] BUONANNO G, JOHNSON G, MORAWSKA L, et al. Volatility characterization of cooking-generated aerosol particles [J]. Aerosol Science and Technology, 2011, 45(9): 1069-1077. doi: 10.1080/02786826.2011.580797 [30] DENNEKAMP M. Ultrafine particles and nitrogen oxides generated by gas and electric cooking [J]. Occupational and Environmental Medicine, 2001, 58(8): 511-516. doi: 10.1136/oem.58.8.511 [31] LI C S, LIN W H, JENQ F T. Size distributions of submicrometer aerosols from cooking [J]. Environment International, 1993, 19(2): 147-154. doi: 10.1016/0160-4120(93)90365-O [32] YEUNG L L, TO W M. Size distributions of the aerosols emitted from commercial cooking processes [J]. Indoor and Built Environment, 2008, 17(3): 220-229. doi: 10.1177/1420326X08092043 [33] MATTI MARICQ M. Chemical characterization of particulate emissions from diesel engines: A review [J]. Journal of Aerosol Science, 2007, 38(11): 1079-1118. doi: 10.1016/j.jaerosci.2007.08.001 [34] RÖNKKÖ T, TIMONEN H. Overview of sources and characteristics of nanoparticles in urban traffic-influenced areas [J]. Journal of Alzheimer's Disease, 2019, 72(1): 15-28. doi: 10.3233/JAD-190170 [35] HALLQUIST M, WENGER J C, BALTENSPERGER U, et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues [J]. Atmospheric Chemistry and Physics, 2009, 9(14): 5155-5236. doi: 10.5194/acp-9-5155-2009 [36] 李庆, 段雷, 蒋靖坤, 等. 我国民用燃煤一次颗粒物的减排潜力研究 [J]. 中国电机工程学报, 2016, 36(16): 4408-4414, 4527. LI Q, DUAN L, JIANG J K, et al. Investigation of reducing potential for primary particulate emission from residential coal combustion in China [J]. Proceedings of the CSEE, 2016, 36(16): 4408-4414, 4527(in Chinese).
[37] LOBO P, DURDINA L, SMALLWOOD G J, et al. Measurement of aircraft engine non-volatile PM emissions: Results of the aviation-particle regulatory instrumentation demonstration experiment (A-PRIDE) 4 campaign [J]. Aerosol Science and Technology, 2015, 49(7): 472-484. doi: 10.1080/02786826.2015.1047012 [38] KUITTINEN N, JALKANEN J-P, ALANEN J, et al. Shipping remains a globally significant source of anthropogenic pn emissions even after 2020 sulfur regulation [J]. Environmental Science & Technology, 2021, 55(1): 129-138. [39] JONSSON Å M, WESTERLUND J, HALLQUIST M. Size-resolved particle emission factors for individual ships [J]. Geophysical Research Letters, 2011, 38(13): L13809. doi: 10.1029/2011gl047672 [40] WANG Z B, HU M, WU Z J, et al. Long-term measurements of particle number size distributions and the relationships with air mass history and source apportionment in the summer of Beijing [J]. Atmospheric Chemistry and Physics, 2013, 13(20): 10159-10170. doi: 10.5194/acp-13-10159-2013 [41] LIU Z R, HU B, ZHANG J K, et al. Characterization of fine particles during the 2014 Asia-Pacific economic cooperation summit: Number concentration, size distribution and sources [J]. Tellus B:Chemical and Physical Meteorology, 2017, 69(1): 1303228. doi: 10.1080/16000889.2017.1303228 [42] ZHU R C, HU J N, BAO X F, et al. Tailpipe emissions from gasoline direct injection (GDI) and port fuel injection (PFI) vehicles at both low and high ambient temperatures [J]. Environmental Pollution, 2016, 216: 223-234. doi: 10.1016/j.envpol.2016.05.066 [43] HARRIS S J, MARICQ M M. Signature size distributions for diesel and gasoline engine exhaust particulate matter [J]. Journal of Aerosol Science, 2001, 32(6): 749-764. doi: 10.1016/S0021-8502(00)00111-7 [44] JETTER J, ZHAO Y X, SMITH K R, et al. Pollutant emissions and energy efficiency under controlled conditions for household biomass cookstoves and implications for metrics useful in setting international test standards [J]. Environmental Science & Technology, 2012, 46(19): 10827-10834. [45] LESKINEN J, TISSARI J, USKI O, et al. Fine particle emissions in three different combustion conditions of a wood chip-fired appliance - Particulate physico-chemical properties and induced cell death [J]. Atmospheric Environment, 2014, 86: 129-139. doi: 10.1016/j.atmosenv.2013.12.012 [46] ROSE EILENBERG S, BILSBACK K R, JOHNSON M, et al. Field measurements of solid-fuel cookstove emissions from uncontrolled cooking in China, Honduras, Uganda, and India [J]. Atmospheric Environment, 2018, 190: 116-125. doi: 10.1016/j.atmosenv.2018.06.041 [47] OBAIDULLAH M, BRAM S, RUYCK J D. An overview of PM formation mechanisms from residential biomass combustion and instruments using in PM measurements[J]. International Journal 0f Energy and Environment, 2018, 12: 326972696. [48] ABDULLAHI K L, DELGADO-SABORIT J M, HARRISON R M. Emissions and indoor concentrations of particulate matter and its specific chemical components from cooking: A review [J]. Atmospheric Environment, 2013, 71: 260-294. doi: 10.1016/j.atmosenv.2013.01.061 [49] KUMAR P, PIRJOLA L, KETZEL M, et al. Nanoparticle emissions from 11 non-vehicle exhaust sources - A review [J]. Atmospheric Environment, 2013, 67: 252-277. doi: 10.1016/j.atmosenv.2012.11.011 [50] 王东滨, 郝吉明, 蒋靖坤. 民用固体燃料燃烧超细颗粒物排放及其潜在健康影响 [J]. 科学通报, 2019, 64(33): 3429-3440. WANG D B, HAO J M, JIANG J K. Ultrafine particle emission and its potential health risk from residential solid fuel combustion [J]. Chinese Science Bulletin, 2019, 64(33): 3429-3440(in Chinese).
[51] LIGHTY J S, VERANTH J M, SAROFIM A F. Combustion aerosols: Factors governing their size and composition and implications to human health [J]. Journal of the Air & Waste Management Association, 2000, 50(9): 1565-1618. [52] MORAWSKA L, ZHANG J. Combustion sources of particles. 1. Health relevance and source signatures [J]. Chemosphere, 2002, 49(9): 1045-1058. doi: 10.1016/S0045-6535(02)00241-2 [53] SCHAUER J J, KLEEMAN M J, CASS G R, et al. Measurement of emissions from air pollution sources. 5. C1–C32 organic compounds from gasoline-powered motor vehicles [J]. Environmental Science & Technology, 2002, 36(6): 1169-1180. [54] SCHAUER J J, ROGGE W F, HILDEMANN L M, et al. Source apportionment of airborne particulate matter using organic compounds as tracers [J]. Atmospheric Environment, 1996, 30(22): 3837-3855. doi: 10.1016/1352-2310(96)00085-4 [55] KLEEMAN M J, SCHAUER J J, CASS G R. Size and composition distribution of fine particulate matter emitted from motor vehicles [J]. Environmental Science & Technology, 2000, 34(7): 1132-1142. [56] HUANG X D, OLMEZ I, ARAS N K, et al. Emissions of trace elements from motor vehicles: Potential marker elements and source composition profile [J]. Atmospheric Environment, 1994, 28(8): 1385-1391. doi: 10.1016/1352-2310(94)90201-1 [57] SIMONEIT B R T. Biomass burning—a review of organic tracers for smoke from incomplete combustion [J]. Applied Geochemistry, 2002, 17(3): 129-162. doi: 10.1016/S0883-2927(01)00061-0 [58] MORAWSKA L, RISTOVSKI Z, JAYARATNE E R, et al. Ambient nano and ultrafine particles from motor vehicle emissions: Characteristics, ambient processing and implications on human exposure [J]. Atmospheric Environment, 2008, 42(35): 8113-8138. doi: 10.1016/j.atmosenv.2008.07.050 [59] ZHU Y F, HINDS W C, KIM S, et al. Study of ultrafine particles near a major highway with heavy-duty diesel traffic [J]. Atmospheric Environment, 2002, 36(27): 4323-4335. doi: 10.1016/S1352-2310(02)00354-0 [60] BARONE T L, ZHU Y F. The morphology of ultrafine particles on and near major freeways [J]. Atmospheric Environment, 2008, 42(28): 6749-6758. doi: 10.1016/j.atmosenv.2008.05.019 [61] ZHAO J, KHALIZOV A, ZHANG R Y, et al. Hydrogen-bonding interaction in molecular complexes and clusters of aerosol nucleation precursors [J]. The Journal of Physical Chemistry A, 2009, 113(4): 680-689. doi: 10.1021/jp806693r [62] SCHOBESBERGER S, JUNNINEN H, BIANCHI F, et al. Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules [J]. PNAS, 2013, 110(43): 17223-17228. doi: 10.1073/pnas.1306973110 [63] CHEN M D, TITCOMBE M, JIANG J K, et al. Acid-base chemical reaction model for nucleation rates in the polluted atmospheric boundary layer [J]. PNAS, 2012, 109(46): 18713-18718. doi: 10.1073/pnas.1210285109 [64] BIANCHI F, PRAPLAN A P, SARNELA N, et al. Insight into acid-base nucleation experiments by comparison of the chemical composition of positive, negative, and neutral clusters [J]. Environmental Science & Technology, 2014, 48(23): 13675-13684. [65] YAO L, GARMASH O, BIANCHI F, et al. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity [J]. Science, 2018, 361(6399): 278-281. doi: 10.1126/science.aao4839 [66] CAI R L, YAN C, YANG D S, et al. Sulfuric acid–amine nucleation in urban Beijing [J]. Atmospheric Chemistry and Physics, 2021, 21(4): 2457-2468. doi: 10.5194/acp-21-2457-2021 [67] DAWSON M L, VARNER M E, PERRAUD V, et al. Simplified mechanism for new particle formation from methanesulfonic acid, amines, and water via experiments and ab initio calculations [J]. PNAS, 2012, 109(46): 18719-18724. doi: 10.1073/pnas.1211878109 [68] WANG M Y, KONG W M, MARTEN R, et al. Rapid growth of new atmospheric particles by nitric acid and ammonia condensation [J]. Nature, 2020, 581(7807): 184-189. doi: 10.1038/s41586-020-2270-4 [69] DONAHUE N M, KROLL J H, PANDIS S N, et al. A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution [J]. Atmospheric Chemistry and Physics, 2012, 12(2): 615-634. doi: 10.5194/acp-12-615-2012 [70] DONAHUE N M, ROBINSON A L, PANDIS S N. Atmospheric organic particulate matter: From smoke to secondary organic aerosol [J]. Atmospheric Environment, 2009, 43(1): 94-106. doi: 10.1016/j.atmosenv.2008.09.055 [71] LI Q, JIANG J K, HAO J M. A review of aerosol nanoparticle formation from ions [J]. KONA Powder and Particle Journal, 2015, 32: 57-74. doi: 10.14356/kona.2015013 [72] HIRSIKKO A, NIEMINEN T, GAGNÉ S, et al. Atmospheric ions and nucleation: A review of observations [J]. Atmospheric Chemistry and Physics, 2011, 11(2): 767-798. doi: 10.5194/acp-11-767-2011 [73] MANNINEN H E, NIEMINEN T, ASMI E, et al. EUCAARI ion spectrometer measurements at 12 European sites – analysis of new particle formation events [J]. Atmospheric Chemistry and Physics, 2010, 10(16): 7907-7927. doi: 10.5194/acp-10-7907-2010 [74] ARNOLD F. Atmospheric ions and aerosol formation [J]. Space Science Reviews, 2008, 137(1/2/3/4): 225-239. [75] KULMALA M, VEHKAMÄKI H, PETÄJÄ T, et al. Formation and growth rates of ultrafine atmospheric particles: A review of observations [J]. Journal of Aerosol Science, 2004, 35(2): 143-176. doi: 10.1016/j.jaerosci.2003.10.003 [76] CAI R L, JIANG J K. A new balance formula to estimate new particle formation rate: Reevaluating the effect of coagulation scavenging [J]. Atmospheric Chemistry and Physics, 2017, 17(20): 12659-12675. doi: 10.5194/acp-17-12659-2017 [77] DENG C J, CAI R L, YAN C, et al. Formation and growth of sub-3 nm particles in megacities: Impact of background aerosols [J]. Faraday Discussions, 2021, 226: 348-363. doi: 10.1039/D0FD00083C [78] CAI R L, YANG D S, FU Y Y, et al. Aerosol surface area concentration: A governing factor in new particle formation in Beijing [J]. Atmospheric Chemistry and Physics, 2017, 17(20): 12327-12340. doi: 10.5194/acp-17-12327-2017 [79] WINKLER P M, ORTEGA J, KARL T, et al. Identification of the biogenic compounds responsible for size-dependent nanoparticle growth [J]. Geophysical Research Letters, 2012, 39(20): 2012GL053253. doi: 10.1029/2012GL053253 [80] EHN M, THORNTON J A, KLEIST E, et al. A large source of low-volatility secondary organic aerosol [J]. Nature, 2014, 506(7489): 476-479. doi: 10.1038/nature13032 [81] MARPLE V A, RUBOW K L, BEHM S M. A microorifice uniform deposit impactor (MOUDI): Description, calibration, and use [J]. Aerosol Science and Technology, 1991, 14(4): 434-446. doi: 10.1080/02786829108959504 [82] MISRA C, KIM S, SHEN S, et al. A high flow rate, very low pressure drop impactor for inertial separation of ultrafine from accumulation mode particles [J]. Journal of Aerosol Science, 2002, 33(5): 735-752. doi: 10.1016/S0021-8502(01)00210-5 [83] SMITH J N, DRAPER D C, CHEE S, et al. Atmospheric clusters to nanoparticles: Recent progress and challenges in closing the gap in chemical composition [J]. Journal of Aerosol Science, 2021, 153: 105733. doi: 10.1016/j.jaerosci.2020.105733 [84] WAGNER A C, BERGEN A, BRILKE S, et al. Size-resolved online chemical analysis of nanoaerosol particles: A thermal desorption differential mobility analyzer coupled to a chemical ionization time-of-flight mass spectrometer [J]. Atmospheric Measurement Techniques, 2018, 11(10): 5489-5506. doi: 10.5194/amt-11-5489-2018 [85] 陈岩, 王炜罡, 刘明元, 等. 纳米颗粒物化学成分测量技术及其应用 [J]. 大气与环境光学学报, 2020, 15(6): 402-412. CHEN Y, WANG W G, LIU M Y, et al. Measurement technologies of nanoparticle chemical composition and their application [J]. Journal of Atmospheric and Environmental Optics, 2020, 15(6): 402-412(in Chinese).
[86] VOISIN D, SMITH J N, SAKURAI H, et al. Thermal desorption chemical ionization mass spectrometer for ultrafine particle chemical composition [J]. Aerosol Science and Technology, 2003, 37(6): 471-475. doi: 10.1080/02786820300959 [87] SMITH J N, MOORE K F, MCMURRY P H, et al. Atmospheric measurements of sub-20 nm diameter particle chemical composition by thermal desorption chemical ionization mass spectrometry [J]. Aerosol Science and Technology, 2004, 38(2): 100-110. doi: 10.1080/02786820490249036 [88] ZHANG R Y, WANG L, KHALIZOV A F, et al. Formation of nanoparticles of blue haze enhanced by anthropogenic pollution [J]. PNAS, 2009, 106(42): 17650-17654. doi: 10.1073/pnas.0910125106 [89] GONSER S G, HELD A. A chemical analyzer for charged ultrafine particles [J]. Atmospheric Measurement Techniques, 2013, 6(9): 2339-2348. doi: 10.5194/amt-6-2339-2013 [90] WANG S Y, JOHNSTON M V. Airborne nanoparticle characterization with a digital ion trap-reflectron time of flight mass spectrometer [J]. International Journal of Mass Spectrometry, 2006, 258(1/2/3): 50-57. [91] LOPEZ-HILFIKER F D, MOHR C, EHN M, et al. A novel method for online analysis of gas and particle composition: Description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO) [J]. Atmospheric Measurement Techniques, 2014, 7(4): 983-1001. doi: 10.5194/amt-7-983-2014 [92] LOPEZ-HILFIKER F D, POSPISILOVA V, HUANG W, et al. An extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF) for online measurement of atmospheric aerosol particles [J]. Atmospheric Measurement Techniques, 2019, 12(9): 4867-4886. doi: 10.5194/amt-12-4867-2019 [93] LEE C P, RIVA M, WANG D Y, et al. Online aerosol chemical characterization by extractive electrospray ionization-ultrahigh-resolution mass spectrometry (EESI-orbitrap) [J]. Environmental Science & Technology, 2020, 54(7): 3871-3880. [94] MASSLING A, STOCK M, WEHNER B, et al. Size segregated water uptake of the urban submicrometer aerosol in Beijing [J]. Atmospheric Environment, 2009, 43(8): 1578-1589. doi: 10.1016/j.atmosenv.2008.06.003 [95] MEIER J, WEHNER B, MASSLING A, et al. Hygroscopic growth of urban aerosol particles in Beijing (China) during wintertime: A comparison of three experimental methods [J]. Atmospheric Chemistry and Physics, 2009, 9(18): 6865-6880. doi: 10.5194/acp-9-6865-2009 [96] ZHAO P S, CHEN Y N, SU J. Size-resolved carbonaceous components and water-soluble ions measurements of ambient aerosol in Beijing [J]. Journal of Environmental Sciences, 2017, 54: 298-313. doi: 10.1016/j.jes.2016.08.027 [97] ZHAO P S, DU X, SU J, et al. Aerosol hygroscopicity based on size-resolved chemical compositions in Beijing [J]. Science of the Total Environment, 2020, 716: 137074. doi: 10.1016/j.scitotenv.2020.137074 [98] SUN K, QU Y, WU Q, et al. Chemical characteristics of size-resolved aerosols in winter in Beijing [J]. Journal of Environmental Sciences, 2014, 26(8): 1641-1650. doi: 10.1016/j.jes.2014.06.004 [99] HERNER J D, AW, GAO, et al. Copyright 2005 air & waste management association size and composition distribution of airborne particulate matter in northern California: I—particulate mass, carbon, and water-soluble ions [J]. Journal of the Air & Waste Management Association, 2005, 55(1): 30-51. [100] CHOW J C, WATSON J G, LOWENTHAL D H, et al. Size-resolved aerosol chemical concentrations at rural and urban sites in Central California, USA [J]. Atmospheric Research, 2008, 90(2/3/4): 243-252. [101] DING X X, KONG L D, DU C T, et al. Long-range and regional transported size-resolved atmospheric aerosols during summertime in urban Shanghai [J]. Science of the Total Environment, 2017, 583: 334-343. doi: 10.1016/j.scitotenv.2017.01.073 [102] CHEN S C, TSAI C J, HUANG C Y, et al. Chemical mass closure and chemical characteristics of ambient ultrafine particles and other PM fractions [J]. Aerosol Science and Technology, 2010, 44(9): 713-723. doi: 10.1080/02786826.2010.486385 [103] CASS G R, HUGHES L A, BHAVE P, et al. The chemical composition of atmospheric ultrafine particles [J]. Philosophical Transactions of the Royal Society of London Series A:Mathematical, Physical and Engineering Sciences, 2000, 358(1775): 2581-2592. doi: 10.1098/rsta.2000.0670 [104] CABADA J C, REES S, TAKAHAMA S, et al. Mass size distributions and size resolved chemical composition of fine particulate matter at the Pittsburgh supersite [J]. Atmospheric Environment, 2004, 38(20): 3127-3141. doi: 10.1016/j.atmosenv.2004.03.004 [105] SMITH J N, MOORE K F, EISELE F L, et al. Chemical composition of atmospheric nanoparticles during nucleation events in Atlanta [J]. Journal of Geophysical Research Atmospheres, 2005, 110(D22): D22S03. [106] SMITH J N, DUNN M J, VANREKEN T M, et al. Chemical composition of atmospheric nanoparticles formed from nucleation in Tecamac, Mexico: Evidence for an important role for organic species in nanoparticle growth [J]. Geophysical Research Letters, 2008, 35(4): L04808. [107] LAWLER M J, RISSANEN M P, EHN M, et al. Evidence for diverse biogeochemical drivers of boreal forest new particle formation [J]. Geophysical Research Letters, 2018, 45(4): 2038-2046. doi: 10.1002/2017GL076394 [108] LAWLER M J, WHITEHEAD J, O'DOWD C, et al. Composition of 15–85 nm particles in marine air [J]. Atmospheric Chemistry and Physics, 2014, 14(21): 11557-11569. doi: 10.5194/acp-14-11557-2014 [109] BZDEK B R, LAWLER M J, HORAN A J, et al. Molecular constraints on particle growth during new particle formation [J]. Geophysical Research Letters, 2014, 41(16): 6045-6054. doi: 10.1002/2014GL060160 [110] SMITH J N, BARSANTI K C, FRIEDLI H R, et al. Observations of aminium salts in atmospheric nanoparticles and possible climatic implications [J]. PNAS, 2010, 107(15): 6634-6639. doi: 10.1073/pnas.0912127107 [111] HODSHIRE A L, LAWLER M J, ZHAO J, et al. Multiple new-particle growth pathways observed at the US DOE Southern Great Plains field site [J]. Atmospheric Chemistry and Physics, 2016, 16(14): 9321-9348. doi: 10.5194/acp-16-9321-2016 [112] HAM W A, KLEEMAN M J. Size-resolved source apportionment of carbonaceous particulate matter in urban and rural sites in central California [J]. Atmospheric Environment, 2011, 45(24): 3988-3995. doi: 10.1016/j.atmosenv.2011.04.063 [113] KLEEMAN M J, RIDDLE S G, ROBERT M A, et al. Source apportionment of fine (PM1. 8) and ultrafine (PM0.1) airborne particulate matter during a severe winter pollution episode [J]. Environmental Science & Technology, 2009, 43(2): 272-279. [114] XUE J, XUE W, SOWLAT M H, et al. Seasonal and annual source appointment of carbonaceous ultrafine particulate matter (PM0.1) in polluted California cities [J]. Environmental Science & Technology, 2019, 53(1): 39-49. [115] GLICKER H S, LAWLER M J, ORTEGA J, et al. Chemical composition of ultrafine aerosol particles in central Amazonia during the wet season [J]. Atmospheric Chemistry and Physics, 2019, 19(20): 13053-13066. doi: 10.5194/acp-19-13053-2019 [116] LAWLER M J, DRAPER D C, SMITH J N. Atmospheric fungal nanoparticle bursts [J]. Science Advances, 2020, 6(3): eaax9051. doi: 10.1126/sciadv.aax9051 [117] BRINES M, DALL'OSTO M, BEDDOWS D C S, et al. Traffic and nucleation events as main sources of ultrafine particles in high-insolation developed world cities [J]. Atmospheric Chemistry and Physics, 2015, 15(10): 5929-5945. doi: 10.5194/acp-15-5929-2015 [118] KONTKANEN J, DENG C J, FU Y Y, et al. Size-resolved particle number emissions in Beijing determined from measured particle size distributions [J]. Atmospheric Chemistry and Physics, 2020, 20(19): 11329-11348. doi: 10.5194/acp-20-11329-2020 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 9005
- HTML全文浏览数: 9005
- PDF下载数: 408
- 施引文献: 0
