

 下载:
下载:

-
砷(As)是一种具有致癌性的重金属元素,自然界与人为产生的砷都造成了环境中砷的聚集,对水体与土壤造成污染,进而对人体造成危害。过往研究表明,无机砷特别是三价砷As(Ⅲ),比五价砷As(Ⅴ)毒性更大,已经被证明是最致癌的物质[1] 。砷污染是目前最普遍的环境问题之一,主要由含砷硫化物氧化形成的酸性矿井水(AMD)引起[2] ,其pH值一般为3或者更低。在中国,高砷土壤主要与采矿活动有关。含砷矿物的氧化溶解,如砷黄铁矿(FeS2 -As)、毒砂(FeAsS)、雄黄(As4S4)、雌黄(As2S3)、钴酸盐(CoAsS),以及它们在多金属矿床和煤层中的副相是矿井水砷的主要来源[3]。富砷硫化物的氧化、富砷铁(氢)氧化物的还原溶解以及铁(氢)氧化物的解吸作用导致地下水砷聚集。
在自然界中,砷通常以无机含氧阴离子形式存在,低pH值环境中以五价砷酸盐H2AsO4−形式、在偏碱性环境中以三价亚砷酸盐H3AsO40形式存在[1],但后者仍然可以大量存在于氧化环境中。在大多数矿井水系中,砷酸盐和亚砷酸盐能够并存于AMD,但长期暴露于大气氧和三价铁矿物表面往往导致砷酸盐物种占优势[3]。比如韩国Ilkwang矿井水中[4],沉淀物矿相中砷以As(Ⅴ)的形式存在。Kinsela等[5]研究了澳大利亚东部以酸性硫酸盐土为主的集水区中砷的存在,发现固态砷几乎只以五价态形式存在于次生三价铁矿物中。
目前常用的含砷废水处理方法可概括为吸附法、化学沉淀法、离子交换法。吸附法是通过物理吸附或化学吸附将污染水中的砷转移到特殊的固体吸附剂中,可用的吸附剂有活性炭、活性铝、黏土、复合铁铝化合物[6]等,各类吸附剂效果不一。化学沉淀法是采用沉淀剂对污染水中的砷进行沉淀过滤,以达到去除的目的,常用的沉淀剂有钙、铁、镁、铝盐以及硫化物等。离子交换法通过离子交换剂中的交换基团与污染水中的有毒阴离子进行交换,进而去除废水中的污染物离子。以上处理方法可以结合使用,其关键在于环境修复材料的选择,不同材料处理效果不一,如Liu等[2]在140 d的柱状实验中用铁尘、泥晶、骨炭及其混合物处理合成含砷AMD,证明3种介质的除砷能力分别为骨炭>泥晶>铁屑。
吸附法操作简便、价格相对低廉,越来越受到众多科学家的重视,是研究应用最多的除砷方法之一。As(Ⅴ)大多存在于氧化环境下,由于其带电性质,很容易发生固相吸附,特别是对铁氧化物、氢化物、羟化物(针铁矿、水铁矿)和羟基硫酸盐(施威特曼石、黄钾铁矾)有较强的被吸附作用[7].
针铁矿、四方针铁矿、赤铁矿、水铁矿和施威特曼石(简称施氏矿物)是土壤、沉积物中的重要组成部分,这些铁矿物对金属和阴离子污染物如砷、铬、铅、汞和硒都有很强的吸附能力[8]。环境中的亚铁离子Fe2+被氧化后,形成的氧化铁氢氧化物(黄钾铁矾、针铁矿、施氏矿物、水铁矿等)能够在其结构中吸收砷或将其吸附在表面,对砷的迁移与修复过程都具有重要的影响。Rodova 等[9]证明0.5 g·dm−3的纳米级零价铁对污染水中的砷有良好的去除作用,单质铁氧化产生氢氧化物时,使As以混合配合物的形式也进入产物结构,从而将溶解的砷从溶液中除去并结合。在被污染的土壤、酸性矿井排水系统、热泉中都发现了砷被固定在含铁矿物中的现象[10]。Farina等[11]在废弃的钨矿和锡矿附近的河岸上收集到受AMD影响的富铁沉淀物,发现其中含有不同比例的施氏矿物、针铁矿和水铁矿颗粒的混合物.
历年来不少研究者对这类潜在的重金属砷吸附材料进行了研究。Perez等[12]进行实验后提出,在缺氧条件下,绿锈GRSO4是去除As(Ⅲ)和As(Ⅴ)的稳定有效的高效吸附剂,并且吸附效率高度依赖pH值,在pH 8—9时对As(Ⅲ)的吸附达到最大值,pH 7时对As(Ⅴ)的吸附达到最大值。Ohnuki等[13]通过对日本群马县一个废弃酸性矿井的污染物排水进行实验,研究了pH 4.7时在一个细菌小环境(生物膜)下对砷的吸收机制,发现由于亚铁离子在生物膜上被氧化形成铁矿物,砷、铁在生物膜上聚集,砷的主要提取机制是砷被吸附在铁硫矿物上或与之共沉淀。Egal等[14]研究了酸性氧化亚铁硫杆菌菌株对富砷酸性矿井排水中次生铁矿物形成的作用,发现在微生物的作用下,形成了施氏矿物、黄钾铁矾、tooeleite等与亚砷酸盐共沉淀。在西班牙南部一项对沙壤和碳酸盐土中污染物离子的研究中发现,砷酸盐在土壤中的保留与铁羟基硫酸盐(黄钾铁矾、施威特曼石)和氧羟化物(针铁矿、水铁矿)的形成有关[15] 。
在众多次生铁矿物中,施氏矿物受到了广泛的关注。施氏矿物结构类似于水铁矿,近30年来对于施氏矿物的研究颇多,众多研究都证明其对砷有着优秀的吸附能力。在高酸性环境且没有其他阴离子竞争作用的情况下,针铁矿和黄钾铁矾对As(Ⅴ)的吸附能力大大低于施氏矿物[16]。水铁矿、针铁矿、施氏矿物和黄钾铁矾在海水中都体现出除砷能力,但在实验条件下,施氏矿物由于酸化海水的缓冲作用释放砷的可能性较小,而针铁矿和黄钾铁矾则出现强烈的砷释放现象,这说明该条件下施氏矿物对砷有更好的固定作用[17]。Raghav等[18]利用砷结晶技术,用臭葱石、砷酸羟磷灰石、砷酸亚铁、陨硫铁、砷酸盐施威特曼石处理含砷固体残渣,发现施氏矿物与砷酸亚铁对砷的固定作用最好。
同时由于施氏矿物的不稳定性,在许多受酸性矿井排水污染的河流中,上游的铁沉积物是施氏矿物,而下游则转化为黄钾铁矾或针铁矿等其他铁矿物,这一转化过程中也可能会造成砷的释放,因此砷的迁移与铁矿物尤其是施氏矿物具有强相关性。施氏矿物作为一种优秀的砷吸附材料受到广泛关注,但该矿物对砷的吸附受到砷形态、pH值、温度等多方因素的影响,在探索富施氏矿物体系中的砷迁移、讨论施氏矿物作为砷污染修复材料的可行性时,研究施氏矿物对砷的吸附机制与效果是一项重要的课题。然而目前缺乏关于施氏矿物对砷的吸附研究的综述。故本文结合国内外研究进展,对施氏矿物的发现、矿物学特征及其与砷的相互作用做了系统性的介绍。
-
施氏矿物(Schwertmannite)是在酸性硫酸盐环境中发现的一种二次铁羟基硫酸盐矿物,在自然环境下广泛形成于富铁和富硫酸盐的酸性介质(比如AMD)中,之后的研究中也逐渐在实验室中被成功制备。由于对污染物的优异吸附能力,施氏矿物可以作为一项优秀的环境修复材料,被用于处理污染水或土壤中的多种重金属,包括砷、铬、磷、铅等。
-
不少研究者发现,砷在矿井排水或者砷富集的河流中出现了衰减,并且在研究中逐渐发现砷的迁移与铁沉淀特别是施氏矿物的形成息息相关。1990年在俄亥俄州东部煤田矿井废水和芬兰一个废弃的铜砷矿污水沉积物样品中,Bigham等[19]发现了一种结晶度差、亚稳态的次生羟基硫酸铁矿物,这种沉积物被认为是在pH 2.5—4.0的富硫酸盐矿井水中赭色沉淀物的主要成分。1994年这种矿物被命名为“施威特曼”(Schwertmannite),以纪念第一个描述施氏矿物的土壤科学家。之后,施氏矿物陆续于1995年、2003年、2006年在澳大利亚、日本、中国被发现[20]。
在矿井水同一地点收集到的沉淀物中发现高浓度的砷,而收集的水样中砷浓度较低,则表明了砷在矿井水中的自然衰减,比如韩国Ilkwang矿井水[4]。在矿区水系中,矿山废弃物中硫化物矿物氧化溶解后释放砷,而大多数被释放的砷通过吸附和共沉淀在下游几米内自然衰减[3]。砷在自然界特别是水域中的迁移与衰减与铁矿物有着重要的关系,一方面铁可以氧化As(Ⅲ),同时铁沉淀吸附As(Ⅴ)对砷的下沉起着重要的作用[21]。As(Ⅲ)流动性更强,因此将自然界中的As(Ⅲ)氧化为As(Ⅴ)也是降低As(Ⅲ)毒性及其流动性的有效途径。
砷在土壤和酸性水体中的移动通过新形成的沉淀(施氏矿物、黄钾铁矾、针铁矿等)的吸附来控制,从而导致砷的自然衰减。Paikaray等[3]也提出矿渣中大部分释放的砷通过与次生铁矿物沉淀(如施氏矿物、针铁矿等)吸附和共沉淀而衰减。Wang等[22]对我国大辽河水系沉积物中进行砷的生态分析发现,沉积物中大部分(62.1%)砷与铁氧化物结合,铁氧化物在浅层环境中对固定大量砷有着重要作用。这些次生铁矿物有效地隔离了砷,降低了溶解砷的含量[23] 。Asta等[24]对西班牙西南部伊比利亚黄铁矿带桑塔罗萨酸流中的沉积物进行的研究发现,砷的迁移过程主要由一些沉淀物(施氏矿物、针铁矿、黄钾铁矾)控制,并且上游沉积的砷主要吸附在施氏矿物上,而下游沉积的砷主要与针铁矿和黄钾铁矾伴生。对法国洛特河流域矿床中砷的空间变异性进行的研究发现[25],河流上游砷浓度很高,而下游砷浓度降低,这种自然衰减与在矿床中富含铁的赭色沉淀物中砷的保留有关;这些沉淀物包括针铁矿和施氏矿物,且上游以施氏矿物为主,下游以针铁矿为主,在上游矿床中观察到施氏矿物向针铁矿的转变,但这种转变似乎并不影响砷在固体中的保留。
在自然环境下,铁硫循环常常被认为与天然施威特曼石的形成与转化有关。施氏矿物已被证明是砷在全球范围内循环的关键角色,但关于砷在土壤中的迁移研究较少而关于其在河流中的迁移研究更多。在韩国Ilkwang[4]矿井水中,通过比较共沉淀与吸附型矿物的X射线吸收近边结构(XANES),发现矿相主要为施氏矿物。砷与施氏矿物共沉淀是该矿井水中砷去除的主要机制,其次是砷被针铁矿吸附,以及砷与水铁矿共沉淀。施氏矿物和水铁矿在酸性矿液中形成,并能够在形成过程中吸收砷,随后施氏矿物矿转化为针铁矿,砷吸附在针铁矿上。
-
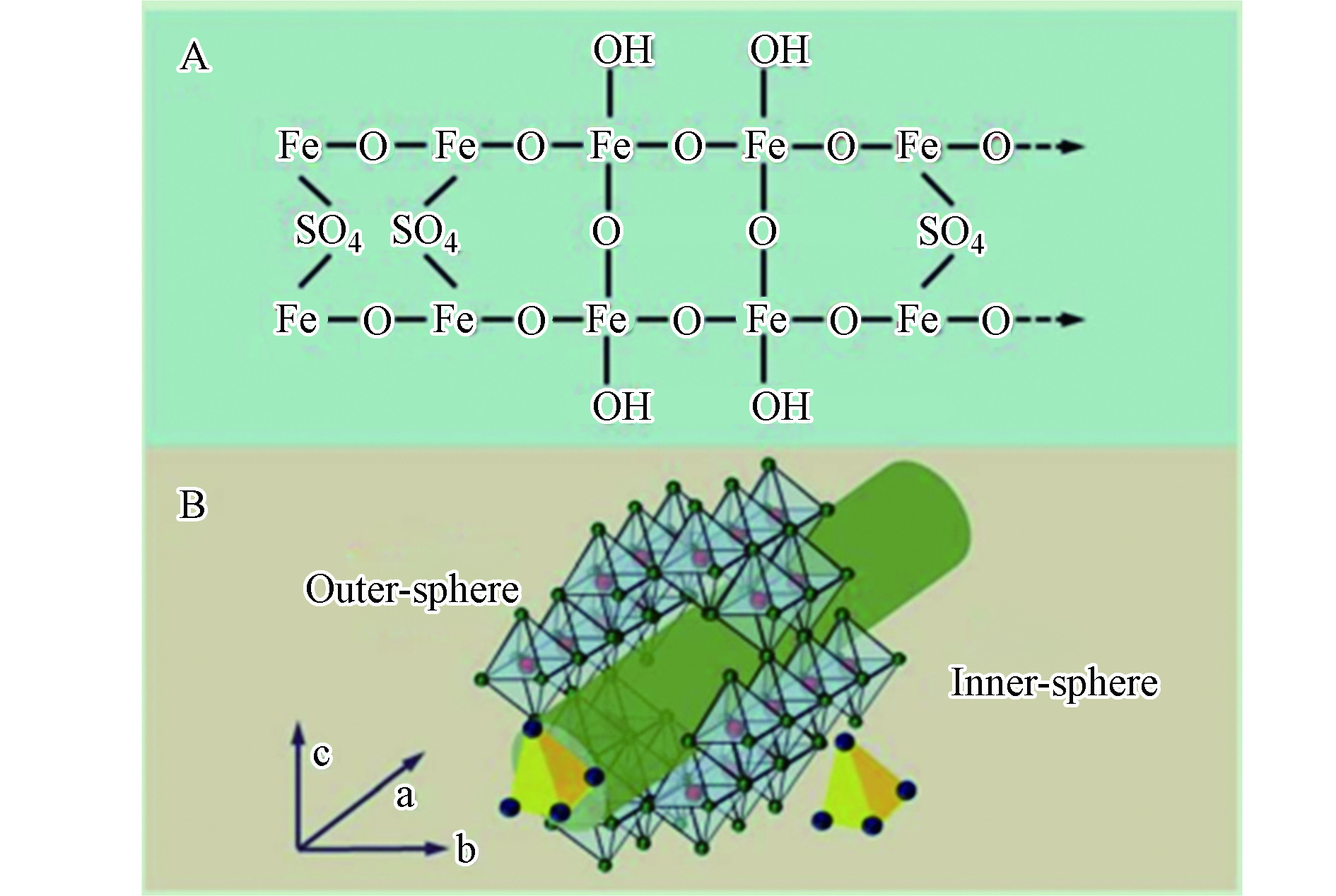
Schwertmann等[26]提出这种矿物的理想化学分子式为Fe8O8(OH)8-2x(SO4)x(1<=x<=1.75),铁硫比为4.6—8.0,其中的Fe完全以三价形式存在,结构内含有大量—OH、—
${\rm{SO}}_4^{2-} $ 基团,其化学组成如图1(A)所示。在理论化学方程式下,其${\rm{SO}}_4^{2-} $ 比率在12.5%—20.5%不等,但不同的合成方法、环境下这个比率可以达到5.3%—32%不等[27]。广泛的研究观察认为,施氏矿物不应该是一种具有重复单元的单晶结构,而是一种多相纳米材料,晶区在无定形质内的宽度小于几纳米[28] 。施氏矿物结晶性比黄钾铁矾和针铁矿差,但相似于水铁矿[29]。Loan等[30]提出施氏矿物的大部分晶须结构与磁赤铁矿一致,都相似于水铁矿,但是还有一些部分同样含有高度无序的水铁矿结构组分和更多的无定形区域,施氏矿物在结晶度上被假定为水铁矿结晶的一种中间态(2线到6线之间)。施氏矿物是一种具有有序通道的三维无序晶体结构,结构示意图如图1(B)。Fukushi等[31]在前人研究的基础上,提出一个可能的配位方式—形似正方针铁矿结构内部存在
${\rm{SO}}_4^{2-} $ 双齿桥。Fernandez-Martinez等[32]提出了一个形似正方针铁矿铁八面体的变形框架,结构中每个晶胞存在的两个硫酸盐分子,在Fe-O八面体构成的通道内形成一个外球和一个內球复合体,孔道内的${\rm{SO}}_4^{2-} $ 很容易被其他相似半径的离子比如砷酸根离子占据[33]。矿物表面和内部都发现了硫酸根,表面络合的${\rm{SO}}_4^{2-} $ 以单齿或单核/双核双齿状存在,而桥联双齿复合体的形式更多出现在在施氏矿物结构通道的内部[34]。在Bigham等[35]的研究中,发现施氏矿物中的硫酸根有1/3吸附在施氏矿物表面,而剩下的2/3存在于通道内。研究发现,合成方式、合成温度等条件的不同都会导致不同的矿物微观形态,施氏矿物的表面形态大体上可分为光滑球状表面和粗糙针垫状或树篱状表面;作为介孔结构的施氏矿物,其比表面积在2—330 m2g−1不等,以微孔和中孔为主[20]。
直接的Fe3+水解是AMD环境中施氏矿物形成的重要途径之一。在Fe3+水解过程中,二聚体(Fe2(OH)24+)出现意味着形成了共边Fe(Ⅲ)聚合物,但硫酸根的存在抑制了二聚体和铁八面体/四面体双角铁键的形成,
${\rm{SO}}_4^{2-} $ 与相邻的铁原子通过Fe—O双角络合,出现Fe(Ⅲ)与${\rm{SO}}_4^{2-} $ 键合的稳定内层络合物[36]。由此,相当多的${\rm{SO}}_4^{2-} $ 内层络合物在中间的FeO6分子团簇上形成,指导施氏矿物的成核与生长[37]。Fe3+水解速率、${\rm{SO}}_4^{2-} $ 浓度、pH、共存离子都影响着水解过程中施氏矿物的形成,矿物形态随水解速率改变,较低的Fe3+水解速率形成较高的矿物结晶度与更好的结构稳定性,同时Cl−不影响施氏矿物的形成,而K+、${\rm{NH}}_4^{+} $ 的存在会略微提高施氏矿物的结晶度[38]。As的存在会阻碍晶须的生长,形成不成熟的施氏矿物[30]。${\rm{SO}}_4^{2-} $ 、Fe3+对次生铁矿物的形成至关重要,只有较低的Fe/S比才能形成施氏矿物[38]。Loan等[30]通过在2线水铁矿的结晶过程中降低过饱和度(在特定的pH值下,过饱和度由铁浓度决定)制备出了部分施氏矿物与2线水铁矿共存,进一步降低Fe3+浓度会导致小比例的针铁矿和赤铁矿结晶,控制Fe3+过饱和度决定了水铁矿与施氏矿物成分的比例。 -
施氏矿物具有不稳定性,可以还原转化为四方硫铁矿和菱铁矿,也可以快速转化为针铁矿,并且释放出硫酸根离子。众多对酸性矿井排水沉积物的研究表明,施氏矿物被还原转化为针铁矿的现象并不少见。比如法国洛特河流域,上游以施氏矿物为主而下游以针铁矿为主。在西班牙南部的力拓与奥迪尔河域[39],阶地沉淀物作为砷的沉淀槽,施氏矿物是受AMD影响的河床中当前形成的主要矿相,而在几个月的时间里这种亚稳态相转变为针铁矿,在较老沉积层的较深处,施氏矿物已部分或全部转化为针铁矿和黄钾铁矾。经过长期转变,古梯田中以结晶良好的针铁矿和成岩赤铁矿为主,且针铁矿中砷的浓度略高于赤铁矿,说明在转化过程中发上了砷的流动。pH值、铁的浓度、有机质、共存离子等都能影响施氏矿物的稳定性,相比之下,实验室制备的施氏矿物由于纯度较高,比天然施氏矿物更为稳定。
影响施氏矿物稳定性的因素诸多,
${\rm{SO}}_4^{2-} $ 与Fe2+对施氏矿物的稳定性至关重要。在一项长达400多天微生物介导还原的施氏矿物培养实验中,Burton等[40]发现铁素体(FeCO3)和四方硫铁矿(FeS)是通过还原施氏矿物生产的主要二次亚铁矿物,并且有25% — 65%的初始施氏矿物也相对快速地转变为针铁矿,转变的程度依赖于${\rm{SO}}_4^{2-} $ 浓度,高浓度${\rm{SO}}_4^{2-} $ 的存在起到了稳定施氏矿物的作用,延缓了其向针铁矿的转变。Fe2+促进施氏矿物溶解并释放${\rm{SO}}_4^{2-} $ ,通过溶解—再结晶机制消耗Fe2+和释放的${\rm{SO}}_4^{2-} $ 形成针铁矿[38]。在接近中性的三价铁Fe(Ⅲ)还原条件下(例如在回流的酸性硫酸盐土壤或矿山湖泊沉积物中),生成的亚铁Fe(Ⅱ)(> 1mmol·L−1的水相Fe(Ⅱ))可以催化施氏矿物向针铁矿的转变[41]。在缺氧碱性条件(pH 8)下,当Fe2+增加时施氏矿物转化形成新矿物的产率加快,而在缺乏Fe2+的情况下,施氏矿物中释放出几乎半数的硫酸根离子[42]。也有研究表明[43],在自然水域中普遍存在的硅元素可以极大地延缓施氏矿物转变的速度和程度,这也是由于硅吸附在施氏矿物表面形成的钝化层可以延缓Fe2+催化其向针铁矿转变的速度,但当Fe2+与施氏矿物反应先发生时,硅的作用将变得微乎其微。因此,在还原条件下的富砷施氏矿物很可能促进砷的移动。高pH值与高温能够促进
${\rm{SO}}_4^{2-} $ 的释放进而促进施氏矿物转变为针铁矿。在pH 2—7范围内,合成施氏矿物对针铁矿的转变率随pH值增加而增加[27]。生物合成施氏矿物在4 ℃的低温下不发生相变,当温度逐渐升高时矿物的相变速率增加[44];在相对低温下,施氏矿物转变为黄钾铁矾与赤铁矿[45],而当加热到400 ℃时会造成施氏矿物热转变为有更大表面积和更小粒径的纳米晶体赤铁矿[46]。共存离子对施氏矿物稳定性的影响不一。Cu2+在Fe2+存在的情况下抑制纯施氏矿物向针铁矿的转变[47],但在酸性条件下(pH3—4),Cu2+对含As(Ⅴ)的施氏矿物的稳定性没有显著影响,而施氏矿物吸附的As(Ⅴ)可以延缓或显著抑制Fe2+催化施氏矿物向针铁矿的转变,但在25 ℃条件下,pH 3—11范围内的As(Ⅴ)-Sch没有发现矿物类变化[48]。结构中As(Ⅴ)的存在阻碍施氏矿物向针铁矿的转变[23],As(Ⅴ)初始浓度增加能减少施氏矿物向针铁矿的转变率[49]。此外,Cl−能够加快
${\rm{SO}}_4^{2-} $ 的释放使施氏矿物转变为针铁矿,但高浓度的Cl−阻碍这一过程[38];K+的存在促进Fe3+水解及之后的沉淀,并且阻碍${\rm{SO}}_4^{2-} $ 的释放,十分有利于形成含K+的聚合球形黄钾铁;高浓度的${\rm{NH}}_4^{+} $ 促进黄钾铁矾的形成,但促进程度远低于K+,并且低浓度的${\rm{NH}}_4^{+} $ 会增加${\rm{SO}}_4^{2-} $ 的释放从而促进施氏矿物向针铁矿的转变。同时,有机质通常也能通过还原和吸附影响施氏矿物的转化,如Jones等[50]研究发现在接近中性的还原条件下富里酸可以显著降低施氏矿物向针铁矿的转变。 -
施氏矿物被发现在酸性环境中,对As(Ⅲ)与As(Ⅴ)都有吸附作用,在碱性环境下也可以吸附As(Ⅲ),但在低pH值条件下施氏矿物对As(Ⅴ)的吸附优于As(Ⅲ),被称作AMD系统中砷的“清道夫”。由于As(Ⅴ)的毒性大于As(Ⅲ),并且在低pH下可溶性更小[51],因此常常将As(Ⅲ)氧化为As(Ⅴ)再去除。由于As(Ⅴ)在酸性矿井排水中更为常见以及其在酸性环境中的高吸附率,目前对施氏矿物吸附As(Ⅴ)的研究较多。
Egal等[14]研究发现在pH 3—4环境下,富As(Ⅲ)AMD中形成的施氏矿物每摩尔Fe对As(Ⅲ)的吸附可以达到300—500 mmol。Burton等[52]在pH 9的条件下同样得到了高达379 mmolAs(Ⅲ)·molFe−1的吸附量,在pH>4.6时,施氏矿物对As(Ⅲ)的吸附优于对As(Ⅴ),但在低砷载荷下,对As(Ⅴ)的吸附优于As(Ⅲ)。Paikaray等[53]、Song等[54]也同样发现施氏矿物在更高的pH值下对As(Ⅲ)的吸附更多,而在低pH值下对As(Ⅴ)的吸附更多。Song等[54]用Fe3+水解合成的施氏矿物吸附As(Ⅴ)和As(Ⅲ),As(Ⅴ)和As(Ⅲ)在200 min内达到吸附平衡,As(Ⅴ)和As(Ⅲ)在pH 3.0时最大吸附量分别为182.86 mg·g−1和45.50 mg·g−1,在pH 7.0时最大吸附量分别为143.25 mg·g−1和217.85 mg·g−1。在这种情况下,酸性环境中As(Ⅲ)的流动性强于As(Ⅴ)。Maillot等[55]研究发现,在酸性水体中沉淀施氏矿物后,As(Ⅲ)的残余溶解度远远高于As(Ⅴ);在AMD系统中,As(Ⅲ)被砷氧化菌如硫单胞菌属催化氧化,As(Ⅲ)的氧化极大推进了富砷AMD系统的环境修复。砷氧化菌微生物活性的季节性变化可能影响自然修复过程,并且在夏季形成更多的As(Ⅴ)[56]。同时,多项研究表明As(Ⅴ)对施氏矿物的成核具有抑制作用[55, 57]。Maillot等[55]也发现As(Ⅲ)对施氏矿物成核后的亲和力远小于As(Ⅴ),在相似铁硫比的固相中,As(Ⅴ)-Sch中溶解As(Ⅴ)浓度比As(Ⅲ)-Sch中溶解As(Ⅲ)浓度低约10倍。
施氏矿物同样表现出良好的再利用能力。Song等[54]用Fe3+水解合成的施氏矿物在7次重复使用后,对As(Ⅴ)和As(Ⅲ)的去除效果甚至更好,在pH 3.0和pH 7.0对As(Ⅴ)的去除率分别为95.3%和63.9%,在pH 3.0和pH 7.0对As(Ⅲ)的去除率分别为31.0%和81.6%。
-
(1)合成方法
天然施氏矿物形成于硫酸根含量丰富的酸性环境中,但天然施氏矿物常常含有不少杂质,为了更好地研究其矿物学特征及环境修复作用,陆续有科学家在实验室中用各种方法人工合成施氏矿物,包括化学氧化法、非生物沉淀法以及生物铁氧化法。其中,化学氧化法更适用于实验中对施氏矿物的理论研究,比如表面形态结构特征等;而缓慢水解与生物方法可以获得吸附效率更高的施氏矿物。
研究表明[58],合成方法对施氏矿物理化性质如比表面积、Fe/S物质的量比、表面形貌、化学成分、可交换硫酸根离子数等有重要影响,而这些对砷的吸附行为有很大的影响。
化学氧化法是简单高效合成施氏矿物的方法之一,包括Fe2+化学氧化和Fe3+非生物沉淀。Fe2+化学氧化法常用的氧化剂有H2O2和MnO2,通常在室温下进行,且获得的施氏矿物粒度细、比表面积相对小。Song等[54]用Fe2+氧化法合成的施氏矿物表面呈球状,比表面积48.2 m2·g−1,而用Fe3+水解合成的施氏矿物表面呈刺状,比表面积325.5 m2·g−1,远远高于前者,并且体现出更优秀的吸附效率。使用H2O2化学氧化法制备施氏矿物时,H2O2的慢速添加导致亚铁离子氧化速度变低、铁沉淀效率更差,但获得的矿物粒子比表面积更大进而具有更强的重金属离子去除效率,并且该法成本十分低廉,计算出的原料成本约为每吨5563 — 9508元[59]。Han等[60]在酸性环境下用MnO2用3种方式氧化Fe(Ⅱ)和As(Ⅲ)——单独氧化、共同氧化、依次氧化Fe(Ⅱ)与As(Ⅲ),不同的氧化方式合成与吸附机制不同。对Fe(Ⅱ)的单独氧化在MnO2表面形成了针状的施氏矿物颗粒,吸附砷酸盐与亚砷酸盐,直到超过吸附上限;在共同氧化时,Fe(Ⅱ)和As(Ⅲ)首先在MnO2上发生竞争性氧化,然后在MnO2颗粒周围形成FeOHAs或FeAsO4,这些FeOHAs和作为FeAsO4的低结晶颗粒悬浮在溶液中,吸附As(Ⅲ)和As(Ⅴ)。
非生物沉淀法通常在高温下进行,包括快速水解与缓慢透析。快速水解法洗涤与分离周期短,但可能导致结构无序等问题。当前研究发现,这种方法与化学氧化法得到的施氏矿物具有相似的粒径与XRD峰值,这可能是由于二者相似的分离方法[20] 。缓慢透析法涉及长时间的透析,并且随后的冷冻干燥过程复杂、昂贵且耗时长[53],但获得的粒径更大,且具有较好的XRD信号以及傅里叶变换红外光谱特征[61]。通常情况下,快速合成的方法形成更光滑的球形表面颗粒,而慢速合成方法常常导致粗糙的针垫表面[20]。合成条件同样会导致施氏矿物针晶结构的显著差异。如快速水解合成施氏矿物时,合成温度的差异影响施氏矿物的表面针的密度,许多研究证明,更高的温度(约85 ℃)通常获得树枝状表面,而更低的温度(<60 ℃)和低pH值(2.4—3.0)条件下时获得的施氏矿物表面针状物更为紧密[20] 。
生物法被认为是一种有效且绿色的方法,近年来受到较多关注与研究,并且早已被应用起来。在自然界中,As(Ⅲ)常常与Fe(Ⅱ)共存,二者可以被共同氧化。酸性矿井排水系统中的Fe2+常常被一类嗜酸微生物氧化,实验室中的生物铁氧化法用细菌或微生物为介质,在室温下氧化Fe2+,比如嗜酸性氧化亚铁硫杆菌(简称A.ferrooxidans)与一种嗜酸的铁氧化菌株[62],前者在自然环境中被发现研究得最多。A.ferrooxidans是一种嗜酸性化学无机营养的革兰氏阴性杆菌,它能够氧化铁矿物盐、亚铁或单质硫作为能量来源。该菌种催化的亚铁氧化反应可以产生大量的氧化铁氢氧化物,如施氏矿物、黄钾铁矾等,因此在存在A.ferrooxidans的自然环境下,铁硫循环常常被与天然施氏矿物的形成与转化有关。2003年对日本西野牧废弃废弃砷矿的研究中发现,在酸性和高SO42-浓度条件下,A.ferrooxidans对亚铁离子的快速催化氧化促进了施氏矿物的形成。
许多科学家都利用微生物在实验室成功获得了施氏矿物。Liao等[63]用A.ferrooxidans反应72 h生成了树篱状的直径约为2.5 μm的施氏矿物颗粒。Duquesne等[64]从酸性矿井排水中分离出一株命名为CC1的酸性氧化亚铁硫杆菌,该菌株只在亚铁上生长时以亚砷酸盐的形式析出砷,并与生成的施氏矿物共沉淀。Xu等[65]从污泥中分离出一种A.菌株HX3,该菌株可以促进纯净施氏矿物的合成,最适合成pH 2.0—3.3,最适合成温度28—35 ℃,可应用于富铁富硫酸盐水环境中砷的去除。李浙英[66]通过A.ferrooxidans生物氧化(生物法)和H2O2氧化水解FeSO4(化学法)制备纯施氏矿物,发现化学法合成矿物速率明显快于生物法,但生物合成施氏矿物颗粒呈现海胆状,而化学合成矿物颗粒成光滑球形,前者粒径与比表面积都更大,合成的施氏矿物吸附性能也更优异。
(2)共存离子
含砷污染物中,通常共存有多种离子,而各类共存离子对施氏矿物除砷效果影响不一。由于砷离子可以吸附在施氏矿物表面或进入其内部,其他共存离子可能会通过与砷离子竞争表面吸附位点或占据矿物内通道来抑制对砷的吸附作用。如Cl−,
${\rm{NO}}_3^{-} $ ,${\rm{CO}}_3^{2-} $ 对其吸附效果没有抑制作用,而${\rm{PO}}_4^{3-} $ 、${\rm{SiO}}_4^{4-} $ 、富里酸(HA)具有干扰性,干扰效果HA>${\rm{PO}}_4^{3-} $ >${\rm{SiO}}_4^{4-} $ [67]。硫酸盐是AMD自然系统中主要的阴离子,由于硫酸盐与砷酸盐竞争表面吸附位置,它的存在对施氏矿物去除砷有负面影响[16]。 Song等[54]实验发现,当硫酸盐浓度从0.2 g·L−1增至6 g·L−1时,施氏矿物对As(Ⅴ)的吸附从85.4 mg·g−1降低到67.6 mg·g−1,但这种影响对As(Ⅲ)的吸附并不明显,同时硝酸盐和氯化物对As(Ⅴ)和As(Ⅲ)的吸附均无明显干扰。除此之外,由于强烈的相互作用,有机共存离子与含磷离子也会影响施氏矿物对砷的去除。Liao等[63]在用生物施氏矿物吸附As(Ⅲ)的实验中同样发现,${\rm{SO}}_4^{2-} $ 与${\rm{PO}}_4^{3-} $ 起到较小程度的抑制作用,但${\rm{SO}}_4^{2-} $ 、${\rm{PO}}_4^{3-} $ 在自然地下水中浓度偏低,因此限制作用十分有限。(3)pH
无论是对As(Ⅲ)还是As(Ⅴ),pH值都严重影响着施氏矿物的吸附作用。在砷浓度不太低时,施氏矿物在低pH条件下对As(Ⅴ)有着高效吸附作用,但吸附作用随着pH值升高而降低;但对于As(Ⅲ),其吸附作用在低pH条件下更为微弱而在高pH环境下增强[52]。随着溶液pH值在3—9范围内增加,生物合成施氏矿物对As(Ⅲ)的吸附增加,As(Ⅲ)的最大去除率发生在pH 7—9左右,最大去除率超过98%,但当溶液pH> 10时,生物型施氏矿物对As(Ⅲ)的吸附急剧下降;因为天然地下水的pH值往往在6—8.5,因此施氏矿物用于去除污染地下水中的As(Ⅲ)时不需要预先调整pH值[63]。
(4)温度
Qiao等[68]以酸性氧化亚铁硫杆菌LX5和溶解的亚铁离子为原料,在培养基中合成了生物施氏矿物,发现105 ℃干燥的0.25 g·L−1施氏矿物对初始浓度为1 mg·L−1 (pH 7.5)的As(Ⅲ)去除率为25.1%,然而,当使用250 ℃加热的施氏矿物作为吸附剂时,去除效率提高到93.0%。在250 ℃的加热条件下处理的施氏矿物有更高的比表面积和更多的吸附位点,获得了最高的吸附效率,说明温度对施氏矿物吸附能力同样有着重要影响。Liao等[63]的实验同样发现生物合成施氏矿物对As(Ⅲ)的吸附能力随温度升高而增加,说明吸附过程是吸热的。
-
在酸性矿山排水和酸性硫酸盐土壤环境中,铁和砷的循环常与施氏矿物的形成和命运联系在一起。施氏矿物对砷的吸附主要有两种方式,一是砷在矿物表面与铁羟基络合,二是砷与矿物表面及结构内部硫酸盐发生配位体交换。
由于施氏矿物内部通道中的
${\rm{SO}}_4^{2-} $ 易被砷替代,当${\rm{AsO}}_3^{3-} $ 与${\rm{AsO}}_4^{3-} $ 被吸附时,大量${\rm{SO}}_4^{2-} $ 被释放出来,从而造成酸度的释放,此类砷酸交换在多个实验中均被发现[58]。${\rm{SO}}_4^{2-} $ 与As(Ⅲ)和As(Ⅴ)都能发生离子交换,且As(Ⅲ)离子交换系数是后者的一半[52]。Paikaray等[58]研究发现1 mol的溶解亚砷酸盐可以有效取代0.12—0.19 mol的施氏矿物中的硫酸根。${\rm{AsO}}_3^{3-} $ 的去除通常有两种方式,直接去除或氧化后再去除[20]。去除机制包括${\rm{SO}}_4^{2-} $ 与${\rm{AsO}}_3^{3-} $ 的离子交换和表面沉淀的形成,后者是主要形式。施氏矿物通过双齿双核与${\rm{AsO}}_3^{3-} $ 结合,表征出特定Fe-As、As-O原子距[69]。施氏矿物对AsO33-的吸附导致${\rm{AsO}}_3^{3-} $ /${\rm{AsO}}_4^{3-} $ -Fe3+-${\rm{SO}}_4^{2-} $ 、${\rm{AsO}}_3^{3-} $ /${\rm{AsO}}_4^{3-} $ -Fe3+型表面沉淀,并在不产生新的矿相的同时造成显著的形态退化。当前最好的${\rm{AsO}}_3^{3-} $ 的两个拟合模型分别是Freundlich 和Langmuir 模型,这两个模型被广泛应用于施氏矿物的吸附研究中,前者表明AsO33-中砷的保留受多层吸附位点控制,而后者则体现出单分子层吸附[20]。许多情况下,施氏矿物会造成AsO33−部分氧化成AsO43-。AsO43−与表面硫酸根和结构硫酸根均可以发生配位体交换,分别在吸附位点形成单配位基或双配位基,但与AsO43−相关的表面活性位点是硫酸根基团而非氢氧根基团[53]。
在过去的研究中,As(Ⅲ)/
${\rm{SO}}_4^{2-} $ 的交换系数可以达到pH 7.5时的0.16[63]与pH 7.1时的0.17[52]。Antelo 等[70]定量研究了不同pH条件下施氏矿物吸附砷酸盐的交换系数。根据他们的研究,只有25%的砷酸盐与结构硫酸盐进行了交换,而其余75%的砷酸盐则吸附在矿物表面的羟基上。 -
多项研究观察发现,施氏矿物对砷的吸附引起矿物衍射图谱与红外光谱的变化,这反应砷吸附机制可以通过离子交换与表面沉淀破坏施氏矿物的形态结构[58]。
Paikaray等[53]发现施氏矿物吸收As(Ⅲ)后,一些典型的衍射峰消失而新的衍射峰出现,这意味着施氏矿物与吸附的As(Ⅲ)的相互作用和可能形成的附加相改变其原有结构,并且改变程度随吸收程度而变化,衍射峰的变化随着As(Ⅲ)加入量的增加而增加。Carlson等[57]和 Regenspurg等[33]在研究施氏矿物吸附As(Ⅴ)的过程中也发现了同样的峰值变化,并推测这是由于形成了铁砷酸盐型表面沉淀物。但Song等[54]通过XRD和TEM(透射电子显微镜)分析研究发现,用Fe3+水解生成的施氏矿物吸附As(Ⅲ)和As(Ⅴ)后其结晶结构和形貌没有明显变化。
同时多项研究表明施氏矿物对砷的吸收延缓其向针铁矿的转变[33,41,71]。 Burton等[41]利用合成的施氏矿物和各种Fe(Ⅱ)、As(Ⅲ)、As(Ⅴ)的添加物进行了一系列9 d的缺氧转化实验。结果发现在无砷条件下,10 m mol·L−1 Fe(Ⅱ)催化施氏矿物快速完全转变为针铁矿,然而,在1 m mol·L−1As(Ⅲ)存在的情况下其催化转化的幅度降至72%,而在1 mmol·L−1As(Ⅴ)存在的情况下,其催化转化的幅度为6%。这说明 As(Ⅲ)与As(Ⅴ)均能抑制施氏矿物的转变,但As(Ⅲ)的效果要弱的多。
-
施氏矿物对砷有着优异的吸附能力,但含砷的施氏矿物也可能会释放出硫酸根和结合的砷[72],铁八面体在一定情况下进一步聚合成更大的、吸附位点更少的单元,使得砷变得容易浸出而释放到水中,这会对酸性硫酸盐土壤和酸性矿井排水系统造成严重的环境危害。
在沿海酸性硫酸盐土中,施氏矿物中酸性和砷的释放,可能在很大程度上受到环境因素的控制,如降雨、排水沟中的平流、地表径流、潮汐冲刷和季节性天气造成的洪水[73]。其中土壤中的有机质是重要影响因素,在酸性与偏中性(pH 4.5、pH 6.5)时一定浓度的富里酸可以延缓施氏矿物酸度的释放,但却大大增强了砷的释放,并且在接近中性条件下释放砷比率更高;而含砷施氏矿物比纯净的施氏矿物更易造成酸度释放,这可能与含砷施氏矿物更大的表面积和矿物表面的砷酸交换有关[72]。无论在紫外光的照射下和黑暗中,柠檬酸都能显著促进含As(Ⅲ)施氏矿物的还原溶解及砷的释放,此时Al3+的存在明显抑制了柠檬酸和紫外光对Sch - As (Ⅲ)的还原溶解[74]。在紫外照射下[75],酒石酸也可显著提高Sch-As(Ⅲ)的还原溶解与总铁、总砷的释放,而在黑暗中,酒石酸促进纯施氏矿物的溶解但对Sch -As(Ⅲ)作用甚微。此外,Al3+的存在促进了酒石酸对Sch - As (Ⅲ)的还原溶解,在紫外光下反应后,溶解的铁主要以Fe2+形式存在,而As (Ⅲ)被氧化为As(Ⅴ),但当柠檬酸消耗完后,Fe3+与As(Ⅴ)又迅速沉淀。
在HoungAloune等[48]的研究中,铜矿中的含砷施氏矿物在pH 3—11、温度25 ℃时长时间没有发生矿物变化,但矿物中砷的释放对pH值有很强的依赖性。pH 2—7时砷的释放可以忽略不计,但在极度酸性或碱性条件下有大量砷的释放。这些表明含砷施氏矿物中砷的释放受环境影响比如pH值,铜离子、亚铁离子浓度而非时间。在Johnston等[46]设计的合成矿物体系中,含As(Ⅴ)施氏矿物的热转变会导致砷的移动性增强,因为较高温度导致结构内的砷表现出表面络合从而产生更强的可交换性,同时高温(如加热> 400 ℃)会造成施氏矿物转变为有更大表面积更小粒径的纳米晶体赤铁矿。之后,Johnston等[76]进一步研究了在富含有机质的土壤中含As(Ⅴ)施氏矿物的在200—800 ℃范围内热解情况,发现在土壤和自然有机质的存在下,进行加热和燃烧能够使施氏矿物中的As(Ⅴ)被还原为As(Ⅲ)(在300—400 ℃左右还原速度最快),提高了砷的流动性,并且使矿物转变为磁赤铁矿和更高温度下的赤铁矿。而在常温的还原条件下,亚铁诱导形成的施氏矿物吸附的As(Ⅴ)并没有被还原成As(Ⅲ)[77]。含砷施氏矿物的还原溶解过程还原过程受到温度、加热时间和有机质含量的综合影响,与Fe(Ⅱ)的形成呈现显著正相关,但在没有土壤有机质的情况下,加热含As(Ⅴ)施氏矿物不会导致As(Ⅴ)或Fe(Ⅲ)的还原,也不会形成磁赤铁矿。
-
施氏矿物由于其对于污染物的突出吸附能力,毫无疑问是一种优秀的环境修复材料,对其生产方式与直接或间接应用的研究众多。
施氏矿物的主要元素是Fe与S,HoungAloune等[47]利用铜矿浸出液成功制造出可有效去除酸性溶液(pH 3—4)中As(Ⅴ)的施氏矿物,因为铜矿浸出液含有大量铁离子与硫酸盐,且Cu2+不影响合成施氏矿物。Tresintsi等[78]针对饮用水砷处理,在连续流公斤级生产反应器中通过FeSO4沉淀合成施氏矿物,这是首次从水处理厂大规模生产和应用的角度,综述了合成参数对含砷氢氧化物吸附效率的影响,该方法应用简单成本低廉,优化反应参数后的吸附剂对As(Ⅴ)有着高效的吸附能力。
由于生物合成施氏矿物具有优秀的吸附能力,并且被认为合成过程绿色环保,材料来源广泛,对生物合成施氏矿物的研究近年来有所增加。Fernandez-Rojo等[79]针对富砷AMD原位处理,提出在连续流生物反应器中利用生物铁氧化法生产施氏矿物,该方法通过形成非晶态砷酸铁除砷,可作为整个AMD处理过程中第一步。Xie等[80]针对模拟含As(Ⅲ)地下水进行了一系列生物型施氏矿物填充柱吸附实验,提出与已报道的其他吸附剂相比,生物施氏矿物具有更大的穿透体积和更大的吸附能力,并且在吸附与再利用中表现出相对稳定的矿相。但在生物合成施氏矿物的过程中,会有部分矿物粘着在反应器壁上,这些粘着矿物的吸附效率远小于悬浮液中的矿物,但若在合成前提前加入适量施氏矿物便可减少矿物粘着量从而提高合成效率[81]。
在生物型施氏矿物对土壤的吸附课题中也进行了一些研究,对于土壤修复而言,施氏矿物应用于处理工艺的一部分。Janneck等[82]介绍了一个利用微生物从褐煤矿井水中合成施氏矿物并利用其从矿井水中除砷的试验厂,生物型施氏矿物与用于微生物氧化的短杆菌属(Brevibacterium sp). YZ-1已被联用处理砷污染土壤[83]。Chai等[84]研究了一种潜在的有效土壤改良剂。该研究利用氧化亚铁酸硫杆菌对施氏矿物进行了生物合成,制备了施氏矿物(SCH)和酸性活化和碱性活化施氏矿物(A-SCH),并对二者在污染土壤中作为固定化剂的潜力进行了评价,结果得出SCH和A-SCH均能有效地在污染土壤中固定砷,且A-SCH的比表面积比SCH高得多,A-SCH的固定效果甚至优于SCH(特别是在低剂量时)。Yang等[83]首次提出联合利用As(Ⅲ)氧化菌和生物合成施氏矿物来处理高砷土壤,从高砷污染土壤中分离得到短杆菌YZ-1用于污染土壤中氧化As(Ⅲ),氧化亚铁硫杆菌氧化硫酸亚铁合成生物型施氏矿物用于固定As(Ⅴ),结果发现该种联合处理方法处理高污染土壤的效果更好,水溶性砷和可提取NaHCO3−总砷的固定化效率分别达到99.3%和82.6%,这为污染土壤开发一种新的绿色修复策略。
施氏矿物的吸附效率受多种因素影响,同样有许多针对合成施氏矿物进行改造以获得更优异的吸附能力的研究。表面形态对施氏矿物吸附能力有着重要影响,Dou等[68]对合成的施氏矿物分别用转鼓造粒法、挤压法和喷涂法制备了不规则形、圆柱形和球形颗粒,从性能和安全性两个方面对三类颗粒进行了系统的As(Ⅴ)去除评价。结果表明总体上圆柱形与不规则颗粒去除饮用水中砷更有优势,不规则颗粒效果最好,这是由于与球形颗粒相比,不规则和圆柱形颗粒具有更大更高的孔隙率、更丰富的孔隙结构和更大的微孔体积。Dey等[85]利用纳米钴铁氧体(CoFe2O4)纳米粒子(CNSh)对合成施氏矿物表面进行了改性,该合成材料对砷的吸附具有较高的pH敏感性,在pH 5.3左右达到最大砷吸附量。实验得出纳米钴铁氧体增强了施氏矿物的磁性能,使其具有较高的吸附效率。李旭伟[86]等对化学合成施氏矿物进行透析,发现不同孔径的透析过程可以显著提高矿物的比表面积或孔体积,透析后的矿物对废水中砷的吸附能力均有提高。
除了直接用于处理富砷废水或土壤,施氏矿物也能在回收其他物质时用于提纯。Pascua等[87]研究了从废弃地热卤水中提取可二次使用的硅酸盐的一个再循环系统,在该系统中,由于添加了石灰(CaO),过量的溶解钙会在回收物中形成亚砷酸盐。与无机吸附剂相比,施氏矿物在选择性地去除溶解的砷同时不会显著降低二氧化硅浓度,因此在将用过的地热卤水引入再循环系统之前使用施氏矿物或许可以增加硅的回收可行性。
施氏矿物同样可与其他物质进行复合形成复合环境修复材料。Ikeda等[88]将施氏矿物1%—50%的悬浮液与纤维原料(含有氧化钙、氧化硅和氧化铝)按1:(0.1—10)的质量比混合制成一种具有透水性的复合材料,用于卫生设施中净化污水。Okido等[89]介绍了一种磁性化学吸收剂,这种吸附剂由磁铁矿微粒组成的核材料和周围沉淀并与核材料化学结合的施氏矿物复合而成,用于去除废水处理中的有毒离子,如铬酸离子、磷酸盐离子、氟离子、砷酸离子、硒酸盐离子、锑酸离子和铀离子。
-
施氏矿物作为一种新型污染物吸附剂,在二十多年来的研究中取得了不错的进展。由于重金属砷的极大危害性以及施氏矿物对砷的优异吸附能力,关于施氏矿物对砷的吸附研究较为广泛。施氏矿物能够通过表面络合与离子交换去除砷,吸附效率高且再利用能力强,并且具有易于制备与成本低的优点,因此被广泛认为是一种优秀的砷污染修复材料。然而,施氏矿物同样具有不稳定性的特点,易于转化为针铁矿或黄钾铁矾,从而影响其利用。此外,施氏矿物的合成方法、处理环境都影响着施氏矿物的除砷效率,并且含砷施氏矿物再释放砷也是一个受关注的问题。因此,以下梳理了关于施氏矿物除砷应用存在的几个问题及可能的研究方向。
(1)迄今为止的研究说明施氏矿物是一种亚稳态无定形铁矿物,易于转变为其他矿相,尤其易于转变为针铁矿,温度、共存离子、有机质等都能影响施氏矿物的转变。施氏矿物作为一种可以长期存在于环境中的潜在修复材料,对于探究增强其稳定性方面仍待进一步讨论。此外,关于其具体的转变机制仍没有清晰的研究。在许多关于河床沉积物的研究中发现了施氏矿物的转变及砷的迁移,但这种转变是否会造成砷的释放仍是不确定的问题。
(2)受环境因素的影响,含砷施氏矿物的还原溶解也可能造成酸和砷的释放,并且其释放收到pH值、温度、有机质、紫外光等的影响。现有研究对含砷施氏矿物的研究有限,含砷施氏矿物的还原溶解机制与影响因素有待进一步研究。
(3)施氏矿物对砷的吸附效率受多种因素影响,近年来对此进行了不少研究,但为了达到更好的利用效果,对制施氏矿物的改造以及对施氏矿物合成方式的改进方面仍有很大的研究空间。
(4)生物型施氏矿物由于吸附效率高、制备方便、成本低、污染小,被认为是一种环保的绿色修复材料,历年来对其研究不少。天然施氏矿物的形成与微生物的作用联系紧密,微生物来源广泛,如何利用自然界的微生物进行施氏矿物的合成利用是一个值得关注的课题。
施氏矿物的矿物学特征及其除砷研究进展
The mineralogical characteristics of schwertmannite and its progress in arsenic removal
-
摘要: 砷污染是全世界突出的环境问题,对污染水中砷的吸附处理受到广泛关注。施氏矿物凭借其优异的吸附性能与简便经济的制备方法,被认为是一种具有重大潜力的除砷材料。本文概述了施氏矿物的发现及其矿物学特征,重点介绍了施氏矿物对砷的去除效果及其影响因素,并分析了施氏矿物的除砷机理及砷的再释放性,同时对施氏矿物的应用研究进行了总结,最后提出了施氏矿物除砷研究中存在的问题及展望。Abstract: Arsenic pollution is a serious environmental problem all over the world and the adsorption treatment of arsenic in contaminated water has attracted wide attention. Because of its excellent adsorption property and simple and economical preparation method, schwertmannite was considered as an arsenic removal material with great potential. In this paper, a review was presented on the discovery and mineralogical characteristics of schwertmannite as well as the removal effect and influencing factors of arsenic by schwertmannite. The mechanism of arsenic removal and arsenic rerelease of schwetmannite were analyzed and the application research of schwertmannite was summarized. At last, the problems and prospects in the study of arsenic removal were put forward.
-

-
[1] SMEDLEY P L, KINNIBURGH D G. A review of the source, behaviour and distribution of arsenic in natural waters [J]. Applied Geochemistry, 2002, 17(5): 517-568. doi: 10.1016/S0883-2927(02)00018-5 [2] LIU J, CHENG H F, ZHAO F H, et al. Effect of reactive bed mineralogy on arsenic retention and permeability of synthetic arsenic-containing acid mine drainage [J]. Journal of Colloid and Interface Science, 2013, 394: 530-538. doi: 10.1016/j.jcis.2012.12.014 [3] PAIKARAY S. Arsenic geochemistry of acid mine drainage [J]. Mine Water And the Environment, 2015, 34(2): 181-196. doi: 10.1007/s10230-014-0286-4 [4] PARK J H, HAN Y S, AHN J S. Comparison of arsenic co-precipitation and adsorption by iron minerals and the mechanism of arsenic natural attenuation in a mine stream [J]. Water Research, 2016, 106: 295-303. doi: 10.1016/j.watres.2016.10.006 [5] KINSELA A S, COLLINS R N, WAITE T D. Speciation and transport of arsenic in an acid sulfate soil-dominated catchment, eastern Australia [J]. Chemosphere, 2011, 82(6): 879-887. doi: 10.1016/j.chemosphere.2010.10.056 [6] 梁美娜, 刘海玲, 朱义年, 等. 复合铁铝氢氧化物的制备及其对水中砷(V)的去除 [J]. 环境科学学报, 2006,26(3): 438-446. doi: 10.3321/j.issn:0253-2468.2006.03.014 LIANG M N, LIU H L, ZHU Y N, et al. Removal of arsenate from water by using the synthetical iron-aluminum hydroxide complexes [J]. Acta Scientiae Circumstantiae, 2006,26(3): 438-446(in Chinese). doi: 10.3321/j.issn:0253-2468.2006.03.014
[7] DIXIT S, HERING J G. Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals:Implications for arsenic mobility [J]. Environ Sci Technol, 2003, 37(18): 4182-4189. doi: 10.1021/es030309t [8] WAYCHUNAS G A, KIM C S, BANFIELD J F. Nanoparticulate iron oxide minerals in soils and sediments: unique properties and contaminant scavenging mechanisms [J]. Journal Of Nanoparticle Research, 2005, 7(4/5): 409-433. [9] RODOVA A, FILIP J, CERNIK M. Arsenic immobilization by nanoscale zero-valent iron [J]. Ecological Chemistry and Engineering S-Chemia I Inzynieria Ekologiczna S, 2015, 22(1): 45-59. [10] MORIN G, CALAS G. Arsenic in soils, mine tailings, and former industrial sites. [J]. Elements, 2006, 2(2): 97-101. doi: 10.2113/gselements.2.2.97 [11] OTERO-FARINA A, GAGO R, ANTELO J, et al. Surface complexation modelling of arsenic and copper immobilization by iron oxide precipitates derived from acid mine drainage [J]. Boletin De La Sociedad Geologica Mexicana, 2015, 67(3): 493-508. doi: 10.18268/BSGM2015v67n3a12 [12] PEREZ J P H, FREEMAN H M, SCHUESSLER J A, et al. The interfacial reactivity of arsenic species with green rust sulfate (GR(SO4)) [J]. Science Of the Total Environment, 2019, 648: 1161-1170. doi: 10.1016/j.scitotenv.2018.08.163 [13] OHNUKI T, SAKAMOTO F, KOZAI N, et al. Mechanisms of arsenic immobilization in a biomat from mine discharge water [J]. Chemical Geology, 2004, 212(3/4): 279-290. [14] EGAL M, CASIOT C, MORIN G, et al. Kinetic control on the formation of tooeleite, schwertmannite and jarosite by Acidithiobacillus ferrooxidans strains in an As(III)-rich acid mine water [J]. Chemical Geology, 2009, 265(3/4): 432-441. [15] GARCIA I, DIEZ M, MARTIN F, et al. Mobility of arsenic and heavy metals in a sandy-loam textured and carbonated soil [J]. Pedosphere, 2009, 19(2): 166-175. doi: 10.1016/S1002-0160(09)60106-5 [16] ASTA M P, CAMA J, MARTINEZ M, et al. Arsenic removal by goethite and jarosite in acidic conditions and its environmental implications [J]. Journal Of Hazardous Materials, 2009, 171(1/3): 965-972. [17] ALARCON R, GAVIRIA J, DOLD B. Liberation of adsorbed and co-precipitated arsenic from jarosite, schwertmannite, ferrihydrite, and goethite in seawater [J]. Minerals, 2014, 4(3): 603-620. doi: 10.3390/min4030603 [18] RAGHAV M, SHAN J L, SAEZ A E, et al. Scoping candidate minerals for stabilization of arsenic-bearing solid residuals [J]. Journal of Hazardous Materials, 2013, 263: 525-532. doi: 10.1016/j.jhazmat.2013.10.009 [19] BIGHAM J M, SCHWERTMANN U, CARLSON L, et al. A poorly crystallized oxyhydroxysulfate of iron formed by bacterial oxidation of Fe(II) in acid mine waters [J]. Geochimica Et Cosmochimica Acta, 1990, 54(10): 2743-2758. doi: 10.1016/0016-7037(90)90009-A [20] ZHANG Z, BI X, LI X T, et al. Schwertmannite: occurrence, properties, synthesis and application in environmental remediation [J]. RSC Advances, 2018, 8(59): 33583-33599. doi: 10.1039/C8RA06025H [21] ASTA M P, AYORA C, ACERO P, et al. Field rates for natural attenuation of arsenic in Tinto Santa Rosa acid mine drainage (SW Spain) [J]. Journal Of Hazardous Materials, 2010, 177(1/3): 1102-1111. [22] WANG S L, WANG P, MEN B, et al. Chemical forms and ecological risk of arsenic in the sediment of the Daliao River System in China [J]. Environmental Monitoring And Assessment, 2012, 184(4): 2237-2245. doi: 10.1007/s10661-011-2113-8 [23] FUKUSHI K, SASAKI M, SATO T, et al. A natural attenuation of arsenic in drainage from an abandoned arsenic mine dump [J]. Applied Geochemistry, 2003, 18(8): 1267-1278. doi: 10.1016/S0883-2927(03)00011-8 [24] ASTA M P, AYORA C, ROMAN-ROSS G, et al. Natural attenuation of arsenic in the Tinto Santa Rosa acid stream (Iberian Pyritic Belt, SW Spain): The role of iron precipitates [J]. Chemical Geology, 2010, 271(1/2): 1-12. [25] COURTIN-NOMADE A, GROSBOIS C, BRIL H, et al. Spatial variability of arsenic in some iron-rich deposits generated by acid mine drainage [J]. Applied Geochemistry, 2005, 20(2): 383-396. doi: 10.1016/j.apgeochem.2004.08.002 [26] SCHWERTMANN U, BIGHAM J M, MURAD E. The first occurrence of schwertmannite in a natural stream environment [J]. European Journal Of Mineralogy, 1995, 7(3): 547-552. doi: 10.1127/ejm/7/3/0547 [27] REGENSPURG S, BRAND A, PEIFFER S. Formation and stability of schwertmannite in acidic mining lakes [J]. Geochimica Et Cosmochimica Acta, 2004, 68(6): 1185-1197. doi: 10.1016/j.gca.2003.07.015 [28] FRENCH R A, CARABALLO M A, KIM B, et al. The enigmatic iron oxyhydroxysulfate nanomineral schwertmannite: Morphology, structure, and composition [J]. American Mineralogist, 2012, 97(8/9): 1469-1482. [29] MURAD E, ROJIK P. Iron mineralogy of mine-drainage precipitates as environmental indicators:Review of current concepts and a case study from the Sokolov Basin, Czech Republic [J]. Clay Minerals, 2005, 40(4): 427-440. doi: 10.1180/0009855054040181 [30] LOAN M, RICHMOND W R, PARKINSON G M. On the crystal growth of nanoscale schwertmannite [J]. Journal of Crystal Growth, 2005, 275(1/2): e1875-e1881. [31] FUKUSHI K, SATO T, YANASE N, et al. Arsenate sorption on schwertmannite [J]. American Mineralogist, 2004, 89(11/12): 1728-1734. [32] FERNANDEZ-MARTINEZ A, TIMON V, ROMAN-ROSS G, et al. The structure of schwertmannite, a nanocrystalline iron oxyhydroxysulfate [J]. American Mineralogist, 2010, 95(8/9): 1312-1322. [33] REGENSPURG S, PEIFFER S. Arsenate and chromate incorporation in schwertmannite [J]. Applied Geochemistry, 2005, 20(6): 1226-1239. doi: 10.1016/j.apgeochem.2004.12.002 [34] TRESINTSI S, SIMEONIDIS K, PLIATSIKAS N, et al. The role of ${\rm{SO}}_4^{2-} $ surface distribution in arsenic removal by iron oxy-hydroxides [J]. Journal Of Solid State Chemistry, 2014, 213: 145-151. doi: 10.1016/j.jssc.2014.02.026[35] BIGHAM J M, SCHWERTMANN U, PFAB G. Influence of pH on mineral speciation in a bioreactor simulating acid mine drainage [J]. Applied Geochemistry, 1996(6): 845-849. [36] COLLINS R N, ROSSO K M, ROSE A L, et al. An in situ XAS study of ferric iron hydrolysis and precipitation in the presence of perchlorate, nitrate, chloride and sulfate [J]. Geochimica Et Cosmochimica Acta, 2016, 177: 150-169. doi: 10.1016/j.gca.2016.01.021 [37] WANG X, GU C, FENG X, et al. Sulfate local coordination environment in schwertmannite [J]. Environ Sci Technol, 2015, 49(17): 10440-10448. doi: 10.1021/acs.est.5b02660 [38] YING H, FENG X, ZHU M, et al. Formation and transformation of schwertmannite through direct Fe3+ hydrolysis under various geochemical conditions[J]. Environmental ence. Nano, 2020. [39] PARVIAINEN A, CRUZ-HERNANDEZ P, PEREZ-LOPEZ R, et al. Raman identification of Fe precipitates and evaluation of As fate during phase transformation in Tinto and Odiel River Basins [J]. Chemical Geology, 2015, 398: 22-31. doi: 10.1016/j.chemgeo.2015.01.022 [40] BURTON E D, JOHNSTON S G, KRAAL P, et al. Sulfate availability drives divergent evolution of arsenic speciation during microbially mediated reductive transformation of schwertmannite [J]. Environmental Science & Technology, 2013, 47(5): 2221-2229. [41] BURTON E D, JOHNSTON S G, WATLING K, et al. Arsenic effects and behavior in association with the Fe(II)-catalyzed transformation of schwertmannite [J]. Environmental Science & Technology, 2010, 44(6): 2016-2021. [42] PAIKARAY S, PEIFFER S. Lepidocrocite formation kinetics from schwertmannite in Fe(II)-rich anoxic alkaline medium [J]. Mine Water And the Environment, 2015, 34(2): 213-222. doi: 10.1007/s10230-014-0309-1 [43] BURTON E D, JOHNSTON S G. Impact of silica on the reductive transformation of schwertmannite and the mobilization of arsenic [J]. Geochimica Et Cosmochimica Acta, 2012, 96: 134-153. doi: 10.1016/j.gca.2012.08.007 [44] 谢越, 周立祥. 酸性环境下生物成因施氏矿物稳定性研究 [J]. 地学前缘, 2011, 18(5): 310-318. XIE Y, ZHOU L X. Stability of biogenic schwertmannite in acidic solution [J]. Earth Science Frontiers, 2011, 18(5): 310-318(in Chinese).
[45] BARHAM, JOSEPH R. Schwertmannite: A unique mineral, contains a replaceable ligand, transforms to jarosites, hematites, and/or basic iron sulfate [J]. Journal of Materials Research, 1997, 12(10): 2751-2758. doi: 10.1557/JMR.1997.0366 [46] JOHNSTON S G, BURTON E D, MOON E M. Arsenic mobilization is enhanced by thermal transformation of schwertmannite [J]. Environmental Science & Technology, 2016, 50(15): 8010-8019. [47] HOUNGALOUNE S, KAWAAI T, HIROYOSHI N, et al. Study on schwertmannite production from copper heap leach solutions and its efficiency in arsenic removal from acidic sulfate solutions [J]. Hydrometallurgy, 2014, 147: 30-40. [48] HOUNGALOUNE S, HIROYOSHI N, ITO M. Stability of As(V)-sorbed schwertmannite under porphyry copper mine conditions [J]. Minerals Engineering, 2015, 74: 51-59. doi: 10.1016/j.mineng.2015.01.003 [49] CRUZ-HERNANDEZ P, PEREZ-LOPEZ R, NIETO J M. Role of arsenic during the aging of acid mine drainage precipitates [J]. Procedia Earth and Planetary Science, 2017, 17: 233-236. doi: 10.1016/j.proeps.2016.12.079 [50] JONES A M, COLLINS R N, ROSE J, et al. The effect of silica and natural organic matter on the Fe(II)-catalysed transformation and reactivity of Fe(III) minerals [J]. Geochimica Et Cosmochimica Acta, 2009, 73(15): 4409-4422. doi: 10.1016/j.gca.2009.04.025 [51] BATTAGLIA-BRUNET F, DICTOR M C, GARRIDO F, et al. An arsenic(III)-oxidizing bacterial population: selection, characterization, and performance in reactors [J]. J Appl Microbiol, 2002, 93(4): 656-667. doi: 10.1046/j.1365-2672.2002.01726.x [52] BURTON E D, BUSH R T, JOHNSTON S G, et al. Sorption of arsenic(V) and arsenic(III) to schwertmannite [J]. Environmental Science & Technology, 2009, 43(24): 9202-9207. [53] PAIKARAY S, GOTTLICHER J, PEIFFER S. Removal of As(III) from acidic waters using schwertmannite: Surface speciation and effect of synthesis pathway [J]. Chemical Geology, 2011, 283(3/4): 134-142. [54] SONG J, JIA S Y, REN H T, et al. Application of a high-surface-area schwertmannite in the removal of arsenate and arsenite [J]. International Journal Of Environmental Science And Technology, 2015, 12(5): 1559-1568. doi: 10.1007/s13762-014-0528-9 [55] MAILLOT F, MORIN G, JUILLOT F, et al. Structure and reactivity of As(III)- and As(V)-rich schwertmannites and amorphous ferric arsenate sulfate from the Carnoules acid mine drainage, France: Comparison with biotic and abiotic model compounds and implications for As remediation [J]. Geochimica Et Cosmochimica Acta, 2013, 104: 310-329. doi: 10.1016/j.gca.2012.11.016 [56] MORIN G, JUILLOT F, CASIOT C, et al. Bacterial formation of tooeleite and mixed Arsenic(III) or Arsenic(V)-Iron(III) gels in the carnoulbs acid mine drainage, France. A XANES, XRD, and SEM study [J]. Environmental Science & Technology, 2003, 37(9): 1705-1712. [57] CARLSON L, BIGHAM J M, SCHWERTMANN U, et al. Scavenging of As from acid mine drainage by schwertmannite and ferrihydrite: a comparison with synthetic analogues [J]. Environ Sci Technol, 2002, 36(8): 1712-1719. doi: 10.1021/es0110271 [58] PAIKARAY S, PEIFFER S. Biotic and abiotic schwertmannites as scavengers for As(III): Mechanisms and effects [J]. Water Air And Soil Pollution, 2012, 223(6): 2933-2942. doi: 10.1007/s11270-012-1077-9 [59] LIU F W, ZHOU J, ZHANG S S, et al. Schwertmannite synthesis through ferrous ion chemical oxidation under different H2O2 supply rates and its removal efficiency for arsenic from contaminated groundwater [J]. PLoS One, 2015, 10(9): 14. [60] HAN X, LI Y L, GU J D. Oxidation of As(III) by MnO2 in the absence and presence of Fe(II) under acidic conditions [J]. Geochimica Et Cosmochimica Acta, 2011, 75(2): 368-379. doi: 10.1016/j.gca.2010.10.010 [61] SUN H, ZHAO F, WU S. Improvement of synthesizing methods and characterization of Schwertmannite [J]. Acta Petrologica et Mineralogica, 2013, 32(6): 1006-1012. [62] MORI J F, LU S, HANDEL M, et al. Schwertmannite formation at cell junctions by a new filament-forming Fe(II)-oxidizing isolate affiliated with the novel genus Acidithrix [J]. Microbiology, 2016, 162(1): 62-71. doi: 10.1099/mic.0.000205 [63] LIAO Y H, LIANG J R, ZHOU L X. Adsorptive removal of As(III) by biogenic schwertmannite from simulated As-contaminated groundwater [J]. Chemosphere, 2011, 83(3): 295-301. doi: 10.1016/j.chemosphere.2010.12.060 [64] DUQUESNE K, LEBRUN S, CASIOT C, et al. Immobilization of arsenite and ferric iron by acidithiobacillus ferrooxidans and its relevance to acid mine drainage [J]. Applied And Environmental Microbiology, 2003, 69(10): 6165-6173. doi: 10.1128/AEM.69.10.6165-6173.2003 [65] XU Y Q, YANG M, YAO T, et al. Isolation, identification and arsenic-resistance of acidithiobacillus ferrooxidans HX3 producing schwertmannite [J]. Journal Of Environmental Sciences, 2014, 26(7): 1463-1470. doi: 10.1016/j.jes.2014.05.012 [66] 李浙英. 化学与生物成因施氏矿物的矿物学特征及其对水中As(III)吸附去除效果的研究[D]. 南京: 南京农业大学, 2010. LI Z Y. Synthesis and pre-treated of Schwertmannite and its efficiency of arsenic removal from simulated[D]. Nanjing Agricultural University, 2010(in Chinese).
[67] DOU X, MOHAN D, PITTMAN C U, JR. Arsenate adsorption on three types of granular schwertmannite [J]. Water Res, 2013, 47(9): 2938-2948. doi: 10.1016/j.watres.2013.01.035 [68] QIAO X X, LIU L L, SHI J, et al. Heating changes bio-schwertmannite microstructure and arsenic(Ⅲ) removal efficiency [J]. Minerals, 2017, 7(1): 14. doi: 10.3390/min7010014 [69] PAIKARAY S, ESSILFIE-DUGHAN J, GOTTLICHER J, et al. Redox stability of As(Ⅲ) on schwertmannite surfaces [J]. Journal of Hazardous Materials, 2014, 265: 208-216. doi: 10.1016/j.jhazmat.2013.11.068 [70] ANTELO J, FIOL S, GONDAR D, et al. Comparison of arsenate, chromate and molybdate binding on schwertmannite: surface adsorption vs anion-exchange [J]. J Colloid Interface Sci, 2012, 386(1): 338-343. doi: 10.1016/j.jcis.2012.07.008 [71] FUKUSHI K, SATO T, YANASE N. Solid-solution reactions in As(Ⅴ) sorption by schwertmannite [J]. Environ Sci Technol, 2003, 37(16): 3581-3586. doi: 10.1021/es026427i [72] VITHANA C L, SULLIVAN L A, BURTON E D, et al. Liberation of acidity and arsenic from schwertmannite: Effect of fulvic acid [J]. Chemical Geology, 2014, 372: 1-11. doi: 10.1016/j.chemgeo.2014.02.012 [73] BURTON E D, BUSH R T, SULLIVAN L A, et al. Mobility of arsenic and selected metals during re-flooding of iron- and organic-rich acid-sulfate soil [J]. Chemical Geology, 2008, 253(1/2): 64-73. [74] ZHANG J, LI Y X, LI W, et al. The synergistic trigger of the reductive dissolution of Schwertmannite-As(Ⅲ) and the release of arsenic from citric acid and UV irradiation [J]. Chemical Geology, 2019, 520: 11-20. doi: 10.1016/j.chemgeo.2019.05.004 [75] ZHANG J, LI W, LI Y, et al. Tartaric acid-induced photoreductive dissolution of schwertmannite loaded with As(Ⅲ) and the release of adsorbed As(III) [J]. Environmental Pollution, 2019, 245: 711-718. doi: 10.1016/j.envpol.2018.11.047 [76] JOHNSTON S G, BENNETT W W, BURTON E D, et al. Rapid arsenic(Ⅴ)-reduction by fire in schwertmannite-rich soil enhances arsenic mobilisation [J]. Geochimica Et Cosmochimica Acta, 2018, 227: 1-18. doi: 10.1016/j.gca.2018.01.031 [77] FAN C, GUO C L, ZENG Y F, et al. The behavior of chromium and arsenic associated with redox transformation of schwertmannite in AMD environment [J]. Chemosphere, 2019, 222: 945-953. doi: 10.1016/j.chemosphere.2019.01.142 [78] TRESINTSI S, SIMEONIDIS K, VOURLIAS G, et al. Kilogram-scale synthesis of iron oxy-hydroxides with improved arsenic removal capacity: Study of Fe(Ⅱ) oxidation-precipitation parameters [J]. Water Research, 2012, 46(16): 5255-5267. doi: 10.1016/j.watres.2012.06.049 [79] FERNANDEZ-ROJO L, HERY M, LE PAPE P, et al. Biological attenuation of arsenic and iron in a continuous flow bioreactor treating acid mine drainage (AMD) [J]. Water Research, 2017, 123: 594-606. doi: 10.1016/j.watres.2017.06.059 [80] XIE Y, ZHOU L X. Arsenite Removal from Simulated Groundwater by Biogenic Schwertmannite: A Column Trial [J]. Pedosphere, 2013, 23(3): 402-408. doi: 10.1016/S1002-0160(13)60032-6 [81] ZHANG J, SHI J, ZHANG S, et al. Schwertmannite Adherence to the Reactor Wall during the Bio-Synthesis Process and Deterioration of Its Structural Characteristics and Arsenic(III) Removal Efficiency [J]. Minerals, 2017, 7(4): 13. [82] JANNECK E, ARNOLD I, KOCH T, et al. Microbial synthesis of schwertmannite from lignite mine water and its utilization for removal of arsenic from mine waters and for production of iron pigments[C]. International Mine Water Association Symposium – Mine Water and Innovative Thinking. 2010. [83] YANG Z H, WU Z J, LIAO Y P, et al. Combination of microbial oxidation and biogenic schwertmannite immobilization: A potential remediation for highly arsenic contaminated soil [J]. Chemosphere, 2017, 181: 1-8. doi: 10.1016/j.chemosphere.2017.04.041 [84] CHAI L Y, TANG J W, LIAO Y P, et al. Biosynthesis of schwertmannite by Acidithiobacillus ferrooxidans and its application in arsenic immobilization in the contaminated soil [J]. Journal of Soils And Sediments, 2016, 16(10): 2430-2438. doi: 10.1007/s11368-016-1449-7 [85] DEY A, SINGH R, PURKAIT M K. Cobalt ferrite nanoparticles aggregated schwertmannite: A novel adsorbent for the efficient removal of arsenic [J]. Journal of Water Process Engineering, 2014, 3: 1-9. doi: 10.1016/j.jwpe.2014.07.002 [86] 李旭伟, 贺静, 张健, 等. 透析对施氏矿物微观结构及其砷吸附能力的影响 [J]. 环境科学学报, 2020, 40(2): 546-553. LI X W, HE J, ZHANG J, et al. Effects of dialysis on the microstructure of schwertmannite and its arsenic removal ability [J]. Acta Scientiae Circumstantiae, 2020, 40(2): 546-553(in Chinese).
[87] PASCUA C S, MINATO M, YOKOYAMA S, et al. Uptake of dissolved arsenic during the retrieval of silica from spent geothermal brine [J]. Geothermics, 2007, 36(3): 230-242. doi: 10.1016/j.geothermics.2007.03.001 [88] IKEDA H, ITO K, SATO T. Composite material for purifying polluted water, contains fibrous raw material and schwertmannite compound in which one portion of sulfate ion is substituted by anion as such as arsenate ion, phosphate ion or silicate ion: Japan, JP2009136795-A[P]. 2009 [89] OKIDO M, BANDO Y, ESKANDARPOUR A, et al. Magnetic chemical absorber used in waste-liquid processing, comprises composite comprising nuclear material consisting of magnetite fine particles, and schwertmannite precipitated around and chemically bonded with nuclear material: WO2008023853-A1[P].2008 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 5610
- HTML全文浏览数: 5610
- PDF下载数: 103
- 施引文献: 0
















































