




-
死海位于以色列、巴勒斯坦和约旦的交界处,东西两岸分别为外约旦高原和犹太山地。约旦河作为死海海水的最主要来源,自北注入死海。从各方面上来说,死海都是一个很独特的地区。首先,它的湖面海拔约为−415 m,是世界地表高度最低的湖泊。其次,它位于沙漠中,降水少而不规律,且蒸发量大,因此死海海水中的盐分浓度非常高,是世界上盐分浓度居第三位的水体。Nissenbaum等[1]于1977年对死海海水各成分的浓度进行了测量,发现氯离子和溴离子的浓度高达220 g·L−1和5 g·L−1,分别是普通海水中的10倍和100倍。同时,死海海水中的金属阳离子(例如镁、钠、钙、钾等)的浓度也均明显高于一般海水。此外,死海还是一个庞大的盐储藏地,其南部有一个大蒸发池,可以持续生产盐以供给周边居民使用及商业用途。死海中部地区南侧的“Dead Sea Works”工厂,即利用高盐度的死海海水,进行碳酸钾、氯、溴、金属镁等物质的生产[2]。
20世纪90年代末,Hebestreit等[3]通过观测最早发现,在死海地区的对流层大气中,午间会出现氧化溴(BrO)浓度的爆发性增长,同时伴随臭氧浓度的迅速降低,即臭氧耗损现象(ozone depletion events, 简称ODEs)的发生。此前, ODEs现象主要在极地地区被观测到,并被认为是由独属于极地地区的化学反应所引起的现象。而在死海地区所观测到的ODEs,一方面提供了不同于极地条件的ODEs的观测数据,可以进一步研究ODEs在不同环境下的产生机理。另一方面也使研究学者们对此前所得出的关于ODEs的研究结论进行重新审视与讨论。例如在观测到死海地区的ODEs之前,研究学者们普遍认为ODEs需依赖于极地寒冷的环境以及独特的稳定边界层条件而存在[4],然而在前文中所提到的Hebestreit等的观测中,死海地区在炎热的夏季正午(约40 ℃)也发生了ODEs[3],该温度比极地ODEs发生时高40—60 ℃ [5]。由此,死海地区逐渐成为了研究ODEs的重点区域之一。
然而,目前并没有较为详尽的综述文章对死海地区ODEs的研究现状进行介绍,因此本文针对该地区ODEs现有的观测、模拟等研究发展进行了归纳总结,并通过与极地ODEs间的比较以突显死海地区ODEs的特点。本文阐述了决定死海ODEs发生及终止的化学反应机理,以及非均相反应和氮氧化物(NOx,NO+NO2)对于该现象的作用。归纳总结了死海地区特殊的环境因素(地形、气象条件等)对于死海ODEs可能造成的影响,以及死海ODEs对于当地环境的反馈作用。本文有助于指明对死海ODEs研究的未来方向,从而弥补对死海ODEs现象总体认识的空白。
-
由于对死海ODEs的研究是基于极地ODEs的已有成果上的进一步探索,因此本文在描述关于死海地区ODEs研究的发展历史时,将从关于极地ODEs的研究开始,进而阐述关于死海地区ODEs的研究进展,并比较两者之间的异同。
-
极地的ODEs是在20世纪80年代初的北美Alaska州被首次观测到[6]。观测期间,该地区的臭氧浓度发生大幅度的下降,在几个小时或几天内从背景浓度(≈40 nmol·mol−1)迅速降低到检出限(≈0.1 nmol·mol−1)以下。该现象即被称为“臭氧耗损现象(ODEs)”[7]。随后,在加拿大北部Alert市的一系列观测中发现该地深冬/早春的大气边界层中同样发生了ODEs[8],从而证实了该现象的存在。
在后续的关于极地ODEs的观测中,Berg等[9]和Sturges [10]均观测到明显高于背景浓度的“可过滤溴(filterable bromine, 简称f-Br)”,可过滤溴主要是观测中可被收集的气态溴类化合物以及含溴的颗粒物的总和,但其具体化学成分并不明确。随后,Barrie等[11]指出,在ODEs期间f-Br与臭氧之间存在强烈的负相关关系(如图1所示),并提出卤素特别是溴元素及其化合物参与了消耗臭氧的一系列化学反应过程,从而导致了极地ODEs的发生。
自20世纪80年代末至今,研究学者们对于卤素尤其是溴及其化合物在ODEs中的作用做了较为详尽的研究[11-14],提出了包含溴原子(Br)和氧化溴(BrO)在内的催化反应循环(Br消耗臭氧生成BrO,BrO发生自反应生成溴分子或溴原子,从而进一步消耗臭氧)来解释臭氧的耗损。
由于活性溴化物(例如Br和BrO)在极地ODEs中的重要性,研究学者们对于它们的来源及其前体物也进行了深入的研究。1988年加拿大Alert市的观测[15]显示,臭氧和三溴甲烷(bromoform, 分子式CHBr3)之间存在明显的负相关关系,而Yokouchi等[16]于1992年的观测也证实了该发现。Barrie等[11]据此提出CHBr3的光解可能是活性溴化物的主要来源。然而,随后Simpson等[17]指出,CHBr3的光解速率较慢,在北极春季大气中的寿命约为100 d,因此,CHBr3光解产生的溴原子相对较少,不可能作为极地边界层ODEs过程中活性溴化物的主要来源。20世纪90年代末,Wennberg、Platt和Lehrer等提出了一种“溴爆炸机制(bromine explosion mechanism)”[12-13,18-20],解释了极地ODEs期间活性溴化物的主要来源。他们指出,被吸收进固态或者液态悬浮气溶胶中的HOBr分子会与气溶胶中的氢离子(H+)以及溴离子(Br−)间发生非均相反应生成溴气分子(Br2),溴气分子进一步被释放至大气中,从而使大气中的总溴量呈指数上升。而这一机制也得到了科学界的普遍认可。
研究学者们还研究了极地ODEs多发生于深冬和初春的原因[4]。这是由于在此期间,极地地区的大气温度通常低于−20 ℃,这种寒冷的空气易于形成较低且更稳定的大气边界层,从而抑制自由大气与边界层内空气的垂直混合,使得高层富含臭氧的空气无法补充进边界层,因此更有利于极地ODEs的发生。其次,与较低纬度的地区相比,极地的光照条件在该段时期内也较为特殊:冬季极地处于极夜状态,化学物质在大气中的寿命进一步延长,从而使参与形成ODEs事件中的光化学反应的前体物(例如Br2和BrCl)发生累积。而当极夜结束春季到来时,太阳辐射会激发ODEs的一系列光化学反应的进行,从而消耗臭氧。
-
在1997年近一个月的观测中,Hebestreit等[3]首次在死海地区观测到ODEs现象,同时他们也首次在极地外地区观测到并非由极地大气输送所形成的高BrO现象。在该次观测中,Hebestreit等[3]通过测量与BrO吸收光谱高度重合的大气吸收光谱密度,在死海地区的边界层中捕捉到了高浓度的BrO。由于BrO主要由溴原子消耗臭氧所产生,是一种典型的可指示ODEs事件发生的示踪剂,因此该观测中所捕捉到的远高于背景浓度的BrO即标志了该地区ODEs的发生。而与极地ODEs的不同之处在于,死海地区ODEs期间所观测到的BrO混合比几乎均可达到50 pmol·mol−1以上,是极地ODEs期间BrO混合比(约为15 pmol·mol−1)的3倍左右 [21]。并且,死海地区发生ODEs所需的时间也只有1—2 h,是极地ODEs发生时间的约十分之一[3]。
目前科学界对于死海地区ODEs事件的成因仍存有争议。此前,有研究学者们指出,观测到的ODEs主要可分为两类,一类是由当地的化学反应剧烈消耗臭氧所形成,而另一类则是由于站点观测到了从其他地区输送而来的低臭氧浓度的气团。Morin等[22]和Jones等[23]的研究指出,由当地化学反应所形成的ODEs的时间尺度大约是1—3 d或是更长,且臭氧浓度的下降较为平缓;而与之相比,气团输送所形成的ODEs则发生迅速,通常在几小时甚至几分钟内就可以观测到臭氧浓度的急剧下降,并伴随有风速和风向的明显变化。由此,研究学者们指出,在死海地区发生的ODEs由于其臭氧浓度下降速率较快,因此更有可能是由低臭氧含量的气团输送所形成。然而,Jacobi等[24]在北极冰冻圈边缘观测到了发生时间不到7 h的由当地化学反应所形成的ODEs,并提出当地化学反应也可能在几小时之内快速形成ODEs。此外,在1997年Hebestreit等的观测中,通过测量臭氧、氮氧化物、一氧化碳、二氧化硫的浓度以及气象参数(例如风速、风向等)的变化[3],证明了观测到的高浓度BrO是由当地化学反应而并非由其他地区的气团输送所形成,并进而指出在死海地区也完全可能发生由当地化学反应所形成的ODEs。目前,科学界普遍认为,死海海面上观测到的ODEs是由当地化学反应所形成,而在死海周边站点所观测到的ODEs则无法确定是由低臭氧浓度气团的输送还是由当地化学反应所产生。
从现有的观测研究来看,死海地区发生的ODEs与极地地区的ODEs在化学机理上类似,都是由卤素化学所形成。然而两者在时间尺度和空间尺度上都存在明显的区别,并且两地的气象和环境条件都相差甚远。因此在进行死海地区ODEs的研究时,可以以极地ODEs和死海地区ODEs的观测数据作为基础,辅以相关的模式研究,并针对两者的不同之处进行深入探索。
-
自发现死海地区的ODEs现象以来,针对该过程中各大气成分的时空分布以及气象和环境等因素所起的作用,研究学者们已进行了多次大型的外场观测研究。而由于BrO与臭氧之间良好的负相关性,在每次观测中均会测定BrO的混合比,以检测ODEs是否发生。而差分吸收光谱技术 (differential optical absorption spectroscopy, 简称为DOAS) [25]则是最常被用来观测BrO的技术,它通过设置持续的光源(例如太阳光)、发射装置以及接收装置,可以测定BrO在UV(紫外线)波段和可见光波段的吸收,从而反演其浓度。在1997年最早发现死海ODEs的观测中,Hebestreit等[3]即是使用DOAS来测定BrO的混合比。该次观测的地点位于将死海分割成南北两部分的水坝上(见图2标星区域),距离其南部的蒸发盐池约21 km。观测时段是1997年5月27日至6月25日。而该次观测中测得的BrO的混合比约为 (86±10) pmol·mol−1.
在同年Matveev等[5]进行的观测中,同样使用了DOAS对BrO进行了测量。同时用于观测的还有高精度的SO2和NO/NO2分析仪、O3和CO监测仪、以及可观测本地气象参数的监测仪。该次观测的站点同样选取在死海的水坝上(见图2标星区域)。图3中展示的是本次观测中所得到的BrO、臭氧和风向随时间的变化,其中阴影部分标示了风向转换为偏南风的观测时段。在该次近一个月的观测中,几乎每日的午间均可发现BrO浓度的升高(见图3中BrO浓度数据);而在BrO浓度升高期间,臭氧的混合比则十分多变,最高可达120 nmol·mol−1,最低则低于检出限(<2 nmol·mol−1,见图3中臭氧浓度数据)。而在观测中发现,几次较为显著的ODEs(6月5日、6月11日、6月12日、6月15日)均发生于风向偏南的时段(见图4中风向数据),而与之相比,在北风条件下,BrO的混合比则低于检出限,而臭氧的混合比也保持在背景浓度60 nmol·mol−1左右。因此,Matveev等[5]指出,死海地区ODEs以及高BrO的现象主要出现在南风条件下。而该结论也与Hebestreit等[3]的观测结论相一致。
为了进一步确定死海地区ODEs的时空特征,Tas等[2,26]于2001年2月开始,在死海周边的3个站点(分别为北部north、中部mid和南部south站点,见图4中标示)对死海的大气成分进行了约19个月的观测(2001年2月至2002年4月,2002年6月至2002年9月),其中各个站点的观测位置和内容如表1所示,其中包括地面观测和高空观测。此外,Tas等还选取了距离死海西部60 km的受死海地区ODEs影响较小的Beer Sheva地区(见图4中标示),作为参考站进行了同期观测。
Tas等对夏季(8月)和冬季(2月)的观测[2,26]进行总结并指出,臭氧在4个地区(北部、中部、南部站点以及参考站点Beer Sheva)的日变化基本类似,主要表现为早上的低值以及午间光化学作用达到最强时的高值。然而,死海地区臭氧的日变化与Beer Sheva地区相比,仍存在一定的差异,主要体现在标准差上。另外,死海地区3个站点的臭氧混合比的标准差均远大于Beer Sheva地区,在午间也均会出现臭氧混合比几乎降为0的现象。相比之下,Beer Sheva地区臭氧混合比的标准差则较小,且午间不存在臭氧混合比低于20 nmol∙mol−1的情况。该臭氧浓度日变化的不同之处指示出死海地区的3个站点均发生了ODEs,而其中以南部站点发生ODEs的频率最高,也最为强烈。此外,他们从同一站点的两个不同月份的观测结果对比中得出,在夏季(8月),死海北部、中部和南部观测点均观测到了ODEs的发生,然而在该年冬季(2月),死海地区中部则并未观测到ODEs;并且在夏季的观测中,死海周边3个站点的标准差明显大于冬季,且臭氧的最低值也相较冬季更低。由此可看出,相较冬季,死海地区的ODEs更易在夏季发生,并且臭氧的耗损也更明显。
该次观测中Tas等[2,26]还在死海北部的Metzoke Dragot地区(见图4中标示),对高空400 m处的BrO进行了近1个月的观测(表1),从而探究死海ODEs的垂直特征。他们发现,在该次观测中,Metzoke Dragot地区只有5 d出现了BrO上升的情况,而这5 d的边界层高度均低于300 m。由此,Tas等得出结论,死海地区边界层高度的变化对于死海地区ODEs的垂直特征有着决定性的影响。
此外,Tas等在该次观测中也对不同季节中风向与ODEs的关系进行了研究[2,26]。该研究首先阐明了死海地区ODEs发生频率的季节变化:即夏季更易发生ODEs,冬季发生ODEs的频率则较低。而3个观测站点(北部、中部及南部)的不同气象数据对比也指示出了风向并非是死海ODEs发生的决定性因素:三地发生ODEs时风向各异,尤其是南部的观测点(south)在夏季的观测中表现为更易在偏北风的情况下发生ODEs,而这也与之前所述的Hebestreit等[3]和Matveev等[5]的结论,即死海ODEs的发生通常出现在南风情况下不一致。对此,笔者们认为,由于在Hebestreit等[3]和Matveev等[5]发现ODEs的两次观测中,正午(即光化学反应以及臭氧耗损达到最强的时段)所探测到的风向几乎均为偏南风,因此并不能据此就把偏南风作为死海ODEs产生的必要条件之一。
Tas等[2,26]除了对大气组分进行观测以外,还给出了死海地区ODEs的更多细节。例如他们还对死海海水中溴离子和氯离子的浓度及其比值Br−/Cl−,以及溴离子的富集指数,即死海海水和一般海水中溴离子的浓度比值Br− (Dead Sea) /Br− (Ocean Water)进行了观测(表2)。结果表明,死海海水中的氯离子浓度比普通海水高一个量级,而溴离子浓度则比普通海水高两个量级;由于普通海水中溴离子和氯离子的浓度比值即Br−/Cl−通常为103量级,因此死海海水中溴离子和氯离子的浓度比值Br−/Cl−约为104量级。此外,由于碳酸盐的存在,普通海水通常呈弱碱性;而死海海水则呈现出明显的酸性。另外,可能由于南部蒸发盐池的存在,死海海水自北向南各离子的浓度均呈现出增大的趋势,其中以溴离子尤为明显,而pH值则呈降低趋势。由于可释放大量溴化物并消耗臭氧的溴爆炸机制需要消耗溴离子,并且更适宜于在pH值较低的环境下发生。因此,拥有高浓度溴离子和较低pH值的死海海水也是最可能发生溴爆炸机制的反应场所之一,这将在后文第4节进行详细讨论。
2016—2017年Shechner等在死海周边对于ODEs的反应产物——挥发性卤代有机物(volatile halogenated organic compounds)进行了观测[27],并试图通过对比不同卤代有机物的浓度,从而更深入地阐明ODEs过程中所发生的化学反应循环,以及探寻ODEs的发生对于大气边界层中各化学物种浓度的影响。Shechner等通过观测得到了与Tas等[2,26]一致的关于ODEs的季节变化规律,即相比于秋冬季,死海周边地区处于春夏季时可以观测到更高浓度的ODEs反应产物,这也代表了该地区夏季发生的ODEs比冬季更为强烈。此外,Shechner等还指出,ODEs期间所观测到的高浓度的卤代有机物可能来源于该地区的盐湖(即死海)以及高含盐量的土壤。
除了上述的对于ODEs的地面观测以外,2012年5月Holla等首次在死海周边地区对ODEs进行了大气边界层特征以及气溶胶、NO2以及BrO浓度分布的垂直观测[28],并指出山谷风和海陆风对于ODEs发生的重要性。与此前的观测结果[2-3,5,26]类似,该次观测中ODEs期间各化学物种浓度的垂直分布也呈现出了显著的日变化。其中5月10日至12日的3 d中,气溶胶、NO2的浓度与BrO的浓度具有较好的负相关性。具体表现为BrO在正午至下午15时左右出现最大值,而同期观测到的气溶胶与NO2的浓度则逐渐降低。然而在观测后期的5月13、14日两天中,NO2与气溶胶的浓度变化则与前3日相比存在较大的差异,并且在这两天内也并未发现BrO的明显生成,即未产生ODEs。而这两天的气象条件与前3日相比也存在较大的不同。为了解释该现象,Holla等[28]针对死海的地形条件提出了一种可能的垂直环流形势,而该环流形势则是由山谷风和海陆风共同作用所形成(具体机理介绍可见后文3.3节)。Holla等指出,该特定的垂直环流形势是ODEs发生的重要条件之一,而过大的强风则会破坏这种垂直环流形势,从而阻碍ODEs的发生。
遗憾的是,由于成本较高以及技术条件的制约,目前关于死海地区ODEs的观测总体仍然偏少,因此对于该现象的认识及其规律的归纳总结仍不全面。此外,关于死海地区ODEs发生时大气垂直特征的观测则更为少见,而这也对相关的模拟研究造成了一定的阻碍。
-
目前对于死海地区ODEs的模拟多为零维和一维的模拟。Sander等自2005年起首先使用零维箱模式CAABA/MECCA模型[29-31],模拟研究了海洋边界层中卤素化学对于臭氧浓度变化的影响。该模式中包括了较为完整的卤素化学反应,且对于卤素化合物参与的非均相反应的处理也更为细致,这也是目前关于死海ODEs的模拟中在化学反应方面处理最为详细全面的模型。Biazar[32]在一维的化学传输模型UAHCTM_1D中加入了溴化学模块,研究了氮氧化物NOx在臭氧的生消过程中所起的作用。比起前述的Sander等的箱模型,Biazar的模型可以显式地考虑大气垂直扩散等物理过程对于死海ODEs的影响,因此可以更为全面地考虑气象因素在ODEs期间所起的作用,模拟结果也更接近实际。Von Glasow等开发了一维MISTRA模型[33-35],用于进行ODEs的模拟。与其他模型相比,MISTRA模型将气溶胶进行更为精细的分类,该模型根据气溶胶粒径大小将其划分为海盐气溶胶(sea salt aerosol)和硫酸盐气溶胶(sulfate aerosol),并进行后续的模拟和计算。因此该模式可以更准确的估计死海所排放出的pH值较低的悬浮气溶胶对于ODEs的影响。
此外,在观测的基础上,Tas等[36-37]也选用了一维的化学传输模型UAHCTM_1D,试图重现出他们的观测中活性溴化物的变化规律[2,26],并以此研究死海地区氮氧化物(NOx)以及非均相反应对溴化学的影响。模拟结果显示NOx浓度升高对于BrO的浓度有两面性作用:在低NOx环境条件下,提升NOx浓度会促进BrO的产生;而在高NOx环境下,NOx的存在则会阻碍BrO的生成。Smoydzin等[38] 选用了MISTRA模型对死海ODEs进行了模拟,该研究中探讨了气象条件(例如逆温层)对死海地区ODEs发生的影响,模拟结果显示在逆温条件下,边界层内的湍流垂直混合受到抑制,ODEs的发生更加快速,且表现的更为显著。Tas等[39]还选用了CAABA/MECCA模型来研究ODEs对大气中气态汞耗损的影响,研究表明在ODEs过程中死海大气中的气态汞也会发生耗损,并被氧化生成二价汞化合物,从而更易进入生态循环。
总体而言,目前关于死海ODEs的模拟研究主要侧重于研究相关的化学反应,而这也有助于我们进一步认识和理解与死海ODEs相关的化学反应机理,并且可以验证观测中所得到的结论。然而,由于缺乏足量的关于死海ODEs的观测数据的支持,目前关于ODEs的许多模拟都是在假设的理想条件下进行,且大量参数都是通过估算和推测来给定,而非基于实际的观测数据,因此模拟的结果与观测结果误差较大。另外,现阶段对于死海地区ODEs的二维和三维模拟还较为匮乏,模拟通常只能侧重于化学反应或者气象要素变化其中一方面的影响,而不能以一个整体进行研究分析。
-
现有研究指出,对流层中ODEs的发生主要归因于溴化学反应循环对于臭氧的消耗作用[4]。同时氮氧化物和其他卤化物(氯、碘等)也可以通过化学反应产生活性溴的暂时储存物,影响溴的循环,进而改变臭氧消耗的速率。以下对各化学物种在ODEs中所起的作用进行分别介绍。
-
自1988年Sturges等[10]和Barrie等[11]在加拿大的Alert市发现臭氧和可过滤溴之间的负相关关系后,研究学者们就一直在持续探索臭氧耗损的基本化学机理以及溴化物在此机理中所扮演的角色。1995年,Wayne等[40]提出造成对流层中臭氧耗损的主要机制是包含BrO的两个化学反应循环。其中第一个反应循环如(R1)所示:
该反应循环首先由溴原子Br与臭氧发生反应生成BrO,随后 BrO会发生自反应,生成两个溴原子或一个溴气分子,溴气分子通过光解产生溴原子,从而进一步消耗臭氧。该反应循环的总体效应是使臭氧不断地被消耗并转化为氧气分子,而溴化物在该循环过程中的总量和性质都没有发生改变,主要起催化剂的作用。
而第二个反应循环的形式则如(R2)所示:
该反应循环由臭氧的光化学反应产物HO2作为触发机制,其与BrO反应生成HOBr,HOBr光解生成活跃的溴原子再与臭氧发生反应,从而造成臭氧的耗损。此外,该反应还可以增大大气中OH和HO2的浓度比值OH/HO2,从而影响该地区大气的氧化性。
然而,(R1)和(R2)这两个反应循环虽然可以在不消耗活性溴化物的情况下降低臭氧浓度,但却无法解释边界层中活性溴化物总体浓度的上升,而这与极地和死海地区ODEs的实际观测中发现的高浓度BrO不一致。因此,在随后的研究中,研究学者们提出了两个非均相反应以解释ODEs过程中活性溴化物如BrO、Br等物种浓度的迅速增加及惰性溴化物如HOBr、HBr的活化过程。其中第一个非均相反应如(R3)所示:
该反应通过消耗一个气态HOBr分子,从液态或固态介质如悬浮气溶胶中激活出储存的溴离子Br−,从而生成溴气分子Br2并释放到大气中。由于该反应的本质是消耗一个气态HOBr分子中的Br原子,但会从储存介质中激活出两个气态Br原子,因此该非均相反应会使大气中活性溴的含量呈指数增长,因而也被称为“溴爆炸机制”[12-13,18-20]。
而另一个非均相反应则是通过BrONO2的水解 [41-43]:
在非均相反应(R4)中,BrO与NO2反应生成的BrONO2会被气溶胶等介质所吸收,并发生水解反应。产生的HOBr可以进一步激活该介质中储存的溴离子(类似于反应R3),从而生成活性溴类并释放至大气中,最终使大气中溴的总量产生大幅增加。由于死海周边地区存在大量工厂等为污染排放源,使得死海大气中NOx的浓度可达到nmol∙mol−1的量级,因此BrO与NO2的反应产物BrONO2的水解反应对于死海地区ODEs及大气中溴总体含量增加的影响也不可忽略。
而模式研究也证实了加入上述的非均相反应后,模拟结果会有明显改变。Tas等[36]利用UAHCTM_1D模型模拟了死海地区ODEs期间BrO的日变化(见图5)。在他们的模拟中,除了包含完整溴化学的标准组外,还设置了缺少非均相反应(R3)或(R4)以及两者都缺少的对照组。结果表明(见图5),标准组的模拟结果(图5d)与观测最为接近。而同时缺少非均相反应(R3)和(R4)的算例模拟结果(图5a)、以及缺少反应(R4)的算例模拟结果(图5b)则与标准组差异较大,从而证明了非均相反应(R4)在死海ODEs中的重要性。Tas等还发现,只缺少反应(R3)的算例模拟结果(图5c)与标准模拟结果类似,Tas等推论造成该结果的原因可能是在模拟中设置的下垫面的溴排放量偏大,因而使大气中活性溴化物的浓度主要由该下垫面的溴排放量所决定,与非均相反应(R3)速率的依赖性则变小,因此导致在模拟中开/关非均相反应(R3)对计算结果的影响不明显。
溴化学对于死海ODEs的作用除了可以消耗臭氧外,溴原子还易与醛类发生如下反应:
该反应中生成的HBr由于溶解度高,容易被气溶胶或海水所吸收,从而终止活性溴类在大气中的循环以及对臭氧的耗损,因此反应(R5)也被认为是ODEs的终止机制之一。
-
在前文提到的可造成臭氧耗损的第一个反应循环(R1)中,BrO+BrO的自反应在大气中存在其他卤素的氧化物的情况下可以被类似的交叉反应所替代:
其中X,Y分别代表不同的卤素原子,如Cl和I。该反应产生的XY分子会快速的光解,从而生成两个活性卤素原子。并且,由于BrO和ClO、BrO和IO的交叉反应速率比BrO的自反应快1—2个量级,因而在死海边界层中发生的ODEs中,其他卤素如氯(Cl)和碘(I)的化合物存在的作用之一即是促进溴的循环,进而加速臭氧的耗损[44-45]。
除此之外,溴爆炸机制(R3)中随着储存介质如气溶胶中溴离子和氯离子浓度比值Br−/Cl−的变化,还会发生下列反应[46]:
该反应生成的BrCl与Br2类似,也可以被排放至大气边界层中,并进一步光解生成Br原子和Cl原子,进而消耗臭氧。此外,BrCl还可以与溴离子(Br−)反应:
生成溴气分子后排放至大气中,从而增大边界层中的溴含量。研究表明,在悬浮气溶胶中的Br−/Cl−比值较低时,反应(R7)占主导地位,释放出的产物以BrCl为主,而在高Br−/Cl−比值的气溶胶中,则更倾向于发生释放溴气的反应(R8)[46]。
而对于碘化物而言,尽管已有模式研究指出,少量的碘化物就可以大幅度增加臭氧耗损的速率[47]。然而,在死海地区现有的观测中并未发现高浓度的碘化物(例如IO等)。并且,相较于溴化学机制,与碘相关的相当一部分的大气化学反应仍不清楚[48]。因此目前对于碘元素及其化合物在死海地区ODEs中影响的研究仍相对较少。
-
由于死海周边工厂的存在,以及当地旅游业的繁荣发展,与极地不同,死海地区受到人为污染源排放的影响较大。其中对ODEs影响最大的人为排放物即是氮氧化物(NOx,NO+NO2)。它可以通过与Br或BrO发生可逆反应,生成BrNO2或BrONO2:
反应(R9)和(R10)可以将活性溴化物例如Br和BrO暂时进行储存,以此调节溴的循环过程,影响ODEs。
然而目前关于NOx对死海ODEs特别是臭氧耗损速率的影响仍不清楚。在1997年的两次观测中,Hebestreit等[3]和Matveev等[5]发现在BrO浓度达到最大值、臭氧耗损最快时,NO2和NOx的浓度也达到最高,从而说明NOx的存在可能会加速溴化物的爆发增长以及相应的臭氧耗损。然而2001年Tas等的观测中则显示,随着BrO浓度的升高,NO2的混合比呈下降趋势[2],而且在BrO浓度达到最高时NO2的混合比达到了最小值,而这也与此前Hebestreit和Matveev等的观测结论恰巧相反。
对此,Tas等[36-37]利用一维模型UAHCTM_1D,讨论了NOx在ODEs中的变化及其作用。Tas等在模拟中将下垫面排放的NO2通量设置为控制变量,而模拟结果则如图6所示。研究发现,当模拟中设置的NO2通量小于实际通量(3.61×1013 molec·(m2∙s)-1)的10倍时,增大NO2通量对BrOx(Br+BrO)的增大有强烈的促进作用;而在10—40倍之间,增大NO2通量对BrOx浓度变化则影响较小;而当模拟中的NO2通量达到实际中NO2通量的40倍或更大时,在模式中增大NO2通量反而会对BrOx的产生起抑制作用。由此,Tas等提出NO2在ODEs中起到了两面性的作用:一方面,在低NOx环境下,NOx可以与BrO反应生成BrONO2,进而通过水解反应产生HOBr,从而促进溴爆炸机制以及后续的臭氧消耗反应,
反应循环(R11a):
(R11b)所表示 :
另一方面,在高NOx环境下,NOx则会作为活性溴的贮存物而存在。在NOx混合比超过某一阈值(在Tas等的模拟中为40倍实际通量)后,过量的NOx会与溴原子反应,从而大量储存活性溴,降低大气中溴的总量,进而阻碍其对臭氧的进一步耗损。而NOx在ODEs中两面性的作用也与近期Zhou等[49]研究得出的NOx对于极地ODEs影响的两面性的结论相一致。
-
死海地区ODEs与极地ODEs的主要差别之一即是其环境因素的不同。而死海地区独特的地理和气象条件同样也是决定当地产生ODEs的关键所在。以下对可能影响死海地区ODEs的环境因素(死海海水特性、蒸发盐池、地理条件等)进行详细的介绍。
-
死海地区由于其独特的高温环境,蒸发量较大,并且其作为约旦河汇入的终点,流入的海盐无法流出而发生积累。因此观测中发现,死海海水表现出不同于普通海水的高盐浓度[1-3,5],其中溴离子的富集指数达到了普通海水的约一百倍(死海北部的富集指数为94,南部富集指数则达到169),同时,死海海水还表现为较为明显的酸性(见前文2.3中说明)。
由于溴爆炸机制(R3)HOBr (g) + H+ (l) + Br− (l) → Br2 (g) + H2O (l) 的进行需要消耗H+和Br−,因此,死海海水的低pH值和高溴离子浓度都有利于溴爆炸机制的发生,因此死海海水也成为了最有可能直接排放活性溴类的主要自然源之一。此外,由于死海自北向南H+和Br−的浓度均呈上升趋势,因此在理论上,死海南部比北部应更容易发生溴爆炸机制,从而释放更大量的活性溴并导致ODEs的加速产生。而这一点在实际观测中也得到了证实[2,26],在观测中不论在哪个季节,死海南部站点观测到的ODEs的发生频率都要明显高于北部站点。
-
基于死海南部存在一个大蒸发盐池,并且观测发现,只有在南风条件下,死海周边站点才可以观测到ODEs,由此Hebestreit等[3]和Matveev等[5]指出死海地区活性溴化物的主要来源可能是其南部的蒸发盐池,还以此解释了观测中死海南部更易发生ODEs的原因。然而,在Tas等在2001至2002年的观测中,死海南部的ODEs却几乎都发生在北风条件下[2,26],这与先前Hebestreit等[3]和Matveev等[5]的推论并不相符。然而在Tas等的观测中同样可以发现,死海南部(即蒸发盐池附近)ODEs的发生频率要明显高于中部和北部,并且臭氧耗损的强度也会更大。由此,笔者们认为死海南部的蒸发盐池可以提供大量的气溶胶作为溴爆炸反应机制的发生场所,从而促进ODEs的发生,然而并不能由此判定死海南部的蒸发盐池就是造成ODEs的最主要原因。目前,死海地区ODEs期间活性溴化物的主要来源尚不明确,有待进一步的研究确定。
-
造成死海ODEs产生的原因之一还可能是由于其特殊的地形和下垫面类型所引起。首先,死海位于海平面以下400 m左右,其东部和西部分别是均超过1000 m高的外约旦高原和犹太山地,其可以有效的阻挡东风和西风。因此在死海地区观测到的盛行风向大多为南风和北风。由于死海南部蒸发盐池的存在,南风可能会带来更高溴含量以及更高气溶胶浓度的大气,从而造成臭氧的耗损。而与之相反,北风则可能会导致死海北部站点ODEs现象的结束。
其次,死海被沙漠所环绕,在高温环境下(尤其是夏季),周边炎热地区的空气团被传输到死海上空,形成小的逆温层。逆温层可以抑制垂直方向上的混合,从而使下垫面释放的活性溴化物在低层大气中产生积累,因此在夏季也更易观测到ODEs;而如果风速增大或在热力作用下逆温层发生破坏,垂直混合则会将边界层中积累的活性溴化物混合至自由大气中,而边界层也会从高层富含臭氧的空气中得到臭氧的补充,从而使ODEs现象结束。为了验证上述观点,Smoydzin等[38]在模式中通过控制海气间的交换以及边界层与自由大气间的垂直交换,对死海地区ODEs进行了模拟研究。模拟结果如图7所示,左图是边界层与自由大气间的垂直混合可以正常进行的模拟结果,右图则是垂直混合被抑制后的模拟结果。可以看出,垂直混合被抑制的情况下,模式可以较好的模拟出死海发生ODEs期间近地面的高BrO以及臭氧的减少。但当垂直混合加强后,边界层内的臭氧迅速得到补充,则ODEs现象终止。然而在Smoydzin等的模拟中并未考虑死海海水和普通海水的不同之处,此外蒸发盐池可能提供的气溶胶也没有考虑在内。除此之外,该模拟虽然可以解释死海南部发生ODEs较多的原因,但却不能很好的解释在同一观测站点中臭氧的日变化。此外,由于死海地区ODEs的高空观测数据的缺乏,该模式研究中关于垂直方向上的模拟结果的正确性也无法得到验证。
如前文所述,死海西侧地中海所形成的海陆风以及山谷风对于ODEs的发生和终止也会产生重要影响[28]。Holla等[28]通过对2012年5月观测所得到的BrO、NO2和气溶胶浓度的日变化进行数据分析,并基于观测期间的气象条件以及后向轨迹的模拟,指出不同的垂直环流形势会强烈影响ODEs的发生及其终止过程。Holla等指出,死海西侧地中海上所形成的海陆风会携带来充足的水汽,从而为ODEs中的非均相反应提供更多的反应场所,促进ODEs的产生。同时,该海陆风还会与山谷风共同作用形成一个垂直方向上的双层环流形势,在该环流结构中的气团可以发生充分的混合和反应,从而更易产生ODEs。这一模型也解释了观测过程中ODEs期间NO2与气溶胶在垂直方向上的分层现象。而在该次观测的后期(5月13日—14日),过于强劲的风破坏了该双层环流形势,因此观测中NO2与气溶胶的垂直分布与12日相比也产生了较为明显的差异。另外,该强风的存在也加强了垂直对流,从而抑制了ODEs的产生或导致ODEs不易被观测到。
-
死海地区的ODEs对周边环境的影响最突出的体现即是对大气中零价汞的耗损上。零价汞即气态汞,易挥发,在大气中的寿命可达两年。气态汞在ODEs期间会被溴化物所氧化,生成二价汞化合物如HgO和HgBr2,从而变得易于沉降并进入土壤和生物圈。而二价汞形成的化合物对人类有毒害神经的作用[50],其进入食物链后经生物的放大作用后对环境和人类都危害极大。
在观测中最早在ODEs中发现明显的气态汞耗损是在极地的加拿大Alert市[50],在该次观测中Schroeder等发现,Alert市春季发生ODEs期间,大气边界层中的气态汞会发生耗损,并转化为寿命更短的汞化物。随后,Moore等[51]、Obrist等[52]和Peleg等[53]观测发现,死海地区的ODEs过程中也存在气态汞的耗损和氧化。图8中展示了Peleg等[53]在死海周边的Ein Bokek站点所观测到的伴随ODEs出现的较为典型的汞耗损现象。从图8可以看出,在BrO达到峰值时,臭氧耗损也最激烈,而此时气态汞的耗损也达到最强。而后续模式研究[39,53]则表明,溴化学即是造成气态汞转化为二价汞气体/颗粒物的最主要原因,而温度则对气态汞的耗损影响不大。
-
本文对死海地区ODEs现象的特征、发现历史、已有模拟和观测研究以及影响因素等进行了综述。死海地区的ODEs与极地ODEs相比,两者具有类似的现象以及相似的机理,但是由于两地环境的不同而表现出不同的特征,这主要体现在ODEs发生的时长、反应温度以及反应场所的差别上。目前关于死海地区ODEs现象中的溴化学反应机理的研究已经较为成熟,两个重要的非均相反应(溴爆炸机制、BrONO2的水解)通过模拟研究已被证明是造成活性溴化物不断释放进入大气边界层并造成臭氧耗损的主要化学机制。同时,由于死海地区靠近人类居住区,易受到污染物排放的影响,因此氮氧化物(NOx)的浓度变化对于死海ODEs也有明显的影响。在关于氮氧化物浓度对于ODEs及BrO浓度影响的敏感性研究中发现,在低NOx排放环境下,增大氮氧化物的排放对于ODEs的产生有促进作用。而与之相比,在高NOx排放环境下,NOx排放量的增加反而会对ODEs的产生起抑制作用。此外,死海地区特殊的环境和气象要素对于死海ODEs产生和终止的重要性也逐渐受到了研究人员的关注,研究发现逆温层会造成大气边界层中垂直湍流混合受限,从而有助于卤化物在边界层内的累积以及死海ODEs的发生。而死海西侧地中海上所形成的湿润的海陆风也会与死海地区的山谷风发生相互作用,从而形成垂直方向上的双层环流形势,进而促进死海ODEs的产生。
但是由于观测技术和实验研究的制约,与臭氧耗损相关的部分化学机理例如与碘相关的大气化学反应尚待完善。同时,与极地类似,死海地区ODEs现象中活性溴化物的源仍未明确;此外,由于测定的技术制约,对于活性溴参与的非均相反应速率仍然无法对其进行精确的估算,而这也对于进一步深入研究ODEs相关的化学反应机理造成了困难。除此以外,位于死海地区的观测仍较为缺乏,这也不利于后续的模拟等研究。目前关于死海地区ODEs的模式研究也较为简单,多为零维和一维的模拟,模拟所讨论的限定条件也较为单一。然而,中纬度地区的气象条件相比极地更为复杂多变,因此想要单纯依靠零维箱模型或一维模型来模拟出死海地区ODEs以及溴化物浓度的变化是相当困难的。
对于死海地区ODEs现象未来的研究方向,首先应进一步加强在该地区的观测活动,组织更多更完善的关于死海ODEs现象的大型外场观测研究,这样一方面可以为模式验证提供大量可靠的观测数据支撑,另一方面也可以为该现象中一些重要的物理化学过程例如可明显影响ODEs现象的非均相反应的速率估算提供更为准确的依据。其次,在数值模式方面,未来可以应用WRF-Chem[54]和CMAQ[55]等三维中尺度气象化学模型对死海ODEs进行更为深入的研究,例如探讨当地化学反应和气团输送对于某次ODEs各自的贡献大小,以及通过数值模式中的敏感性分析,来回答例如南部蒸发盐池的气溶胶排放对于ODEs期间死海大气中活性溴化物的贡献等科学问题。另外,在未来的后续研究中,研究学者们还应更关注死海地区特殊的气象条件和地形条件对于ODEs现象的影响,以及该现象对于该地区大气中其他成分、化学反应以及当地环境(土壤、河流湖泊等)的影响。而这些关于死海地区ODEs的研究成果也可以为已经观测到ODEs的部分中纬度地区,例如库奇兰恩盐沼[56]等提供良好的研究基础以及参考依据。
关于死海地区臭氧耗损现象(ODEs)的研究综述
A review of ozone depletion events (ODEs) in the area of Dead Sea
-
摘要: 自1997年在死海地区的大气边界层中观测到午间臭氧浓度迅速下降的现象后,研究学者们即对该地区的臭氧耗损现象(ozone depletion events, 简称ODEs)展开了研究。而在此之前,大气边界层内的ODEs现象普遍被认为只会发生在极地地区的特殊大气现象。本文综述了关于死海地区ODEs研究的发展历史,主要展示了促使死海ODEs形成的物理化学机理、关于死海地区ODEs的观测和模拟研究,以及死海地区臭氧耗损现象的特点。与极地ODEs类似,造成死海地区的ODEs过程中臭氧耗损的主要化学物质是活性溴化物如氧化溴(BrO)等。但由于死海地区特殊的环境条件,该地区的ODEs在时间尺度上和空间尺度上与极地ODEs存在明显的差异。目前关于死海ODEs的观测研究主要以地面观测为主,并显示相较冬季,死海夏季ODEs的发生频率更高,臭氧耗损也更为明显。而目前关于死海ODEs的模拟研究则以零维和一维模拟为主,模拟中证实了臭氧和BrO之间的负相关关系,并揭示出非均相反应(溴爆炸机制和BrONO2的水解)对于ODEs的重要影响。另外,模拟中还指出了氮氧化物对于ODEs的两面性作用,以及气象条件(如逆温层)对于ODEs产生的影响。现有研究还发现,死海地区特殊的地形、环境条件及其海水中的高溴含量都是促使当地产生ODEs的关键因素。死海ODEs的发生不仅会改变该地区大气边界层中成分(例如汞)的浓度和寿命,也会影响大气的氧化能力,而这些都会对该地区人类的生活和健康产生潜在的作用,因此有必要加深对于该现象的总体认识。Abstract: Ozone depletion events (ODEs) were found to occur in the atmospheric boundary layer of the Dead Sea area at noon since the year 1997, while before that this phenomenon was recognized occurring mostly in high latitudes such as Arctic. In this paper, we reviewed the study history of the ODEs at the Dead Sea, especially summarizing previous observations and model simulations of this phenomenon. We also described the properties of the ODEs at the Dead Sea and the related physico-chemical processes. It was found in previous studies that similar to ODEs occuring in Arctic, reactive bromine species (such as BrO) also play a key role in the occurrence of ODEs at the Dead Sea. However, due to the unique topographic and environmental condition around the Dead Sea, ODEs in the Dead Sea region behave significantly different from those in Arctic, both in spatial and temporal distribution. The observations of the ODEs in the Dead Sea area were mostly ground-based and showed that the ODEs occur in summer more frequently than in winter. The accompanied ozone decrease was also found stronger in summer. The model simulations of ODEs in the Dead Sea area were mostly zero- or one-dimensional, and help to confirm the anticorrelation between ozone and BrO in the observations of the ODEs. The simulation results also revealed the importance of the heterogeneous reactions (such as the “bromine explosion mechanism” and the hydrolysis of BrONO2) in the occurrence of ODEs. Moreover, the model results discovered the two-sided influence of nitrogen oxides on the destruction of ozone during the ODEs, and the impacts of meteorological conditions such as the temperature inversion on ODEs. It was found that the unique environmental conditions, topography and high bromide concentration in the water of Dead Sea are partly responsible for the occurrence and the termination of the ODEs. The occurrence of ODEs is able to alter the oxidation capacity of the atmosphere as well as the lifetime of many components in the boundary layer of the Dead Sea region. Therefore, ODEs in the area of the Dead Sea would potentially have a large influence on the daily life of human beings nearby, thus requiring a more thorough and deeper knowledge of this phenomenon.
-
Key words:
- ozone /
- ozone depletion events /
- Dead Sea /
- bromine /
- halogen /
- nitrogen oxides
-

-
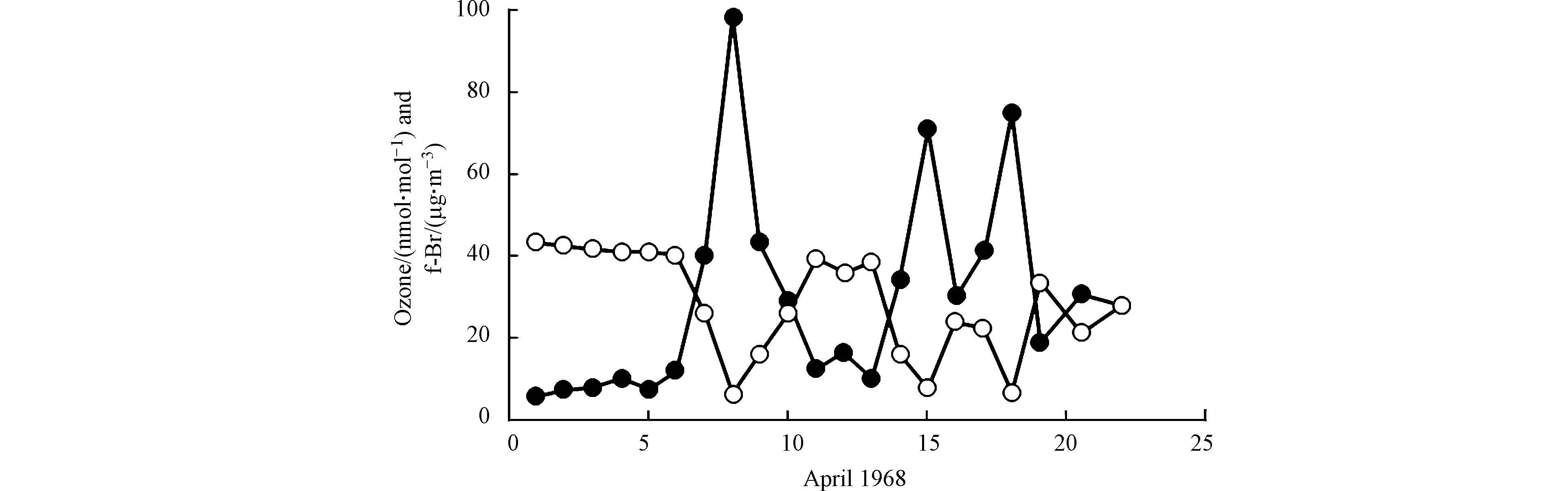
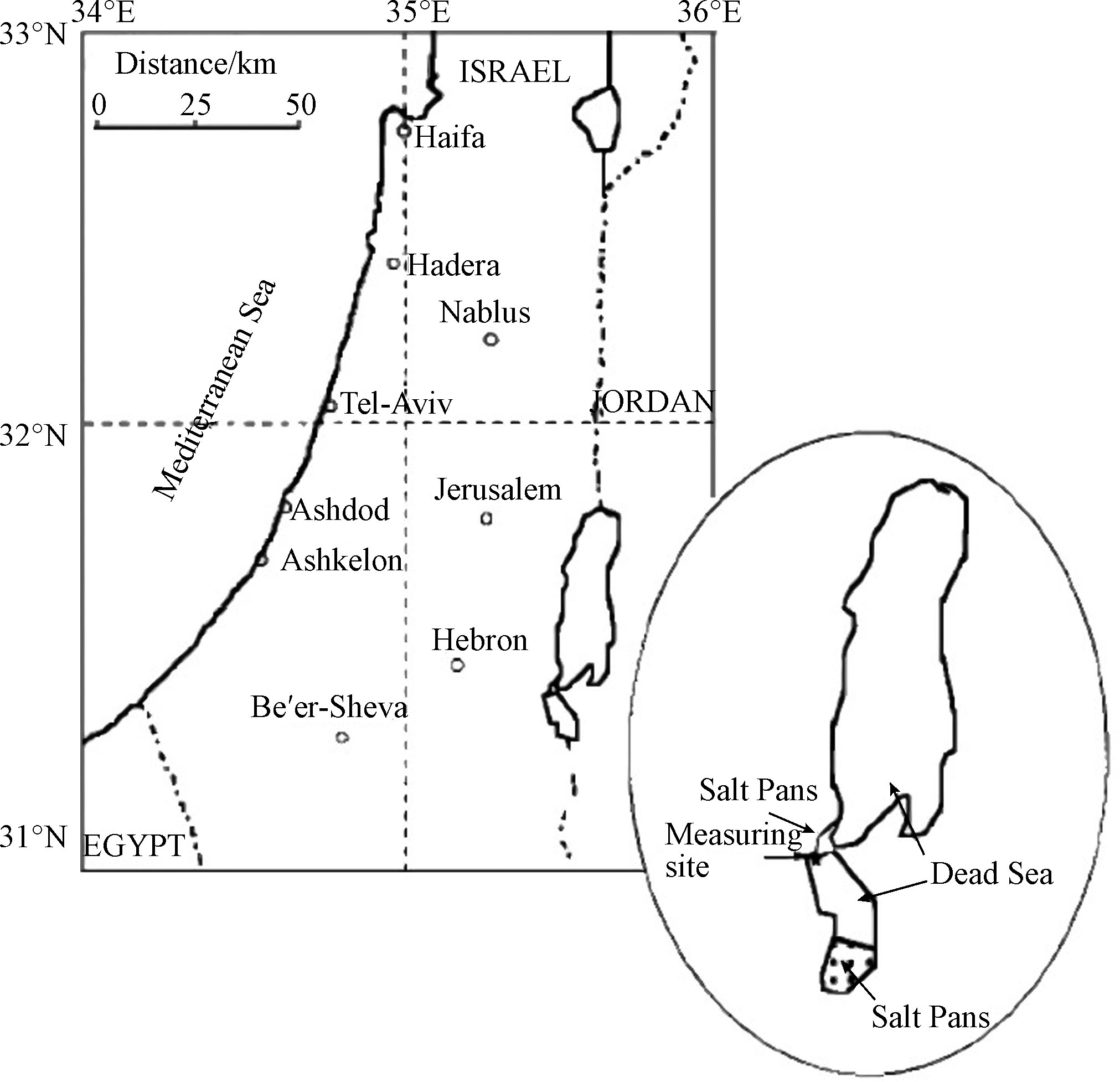
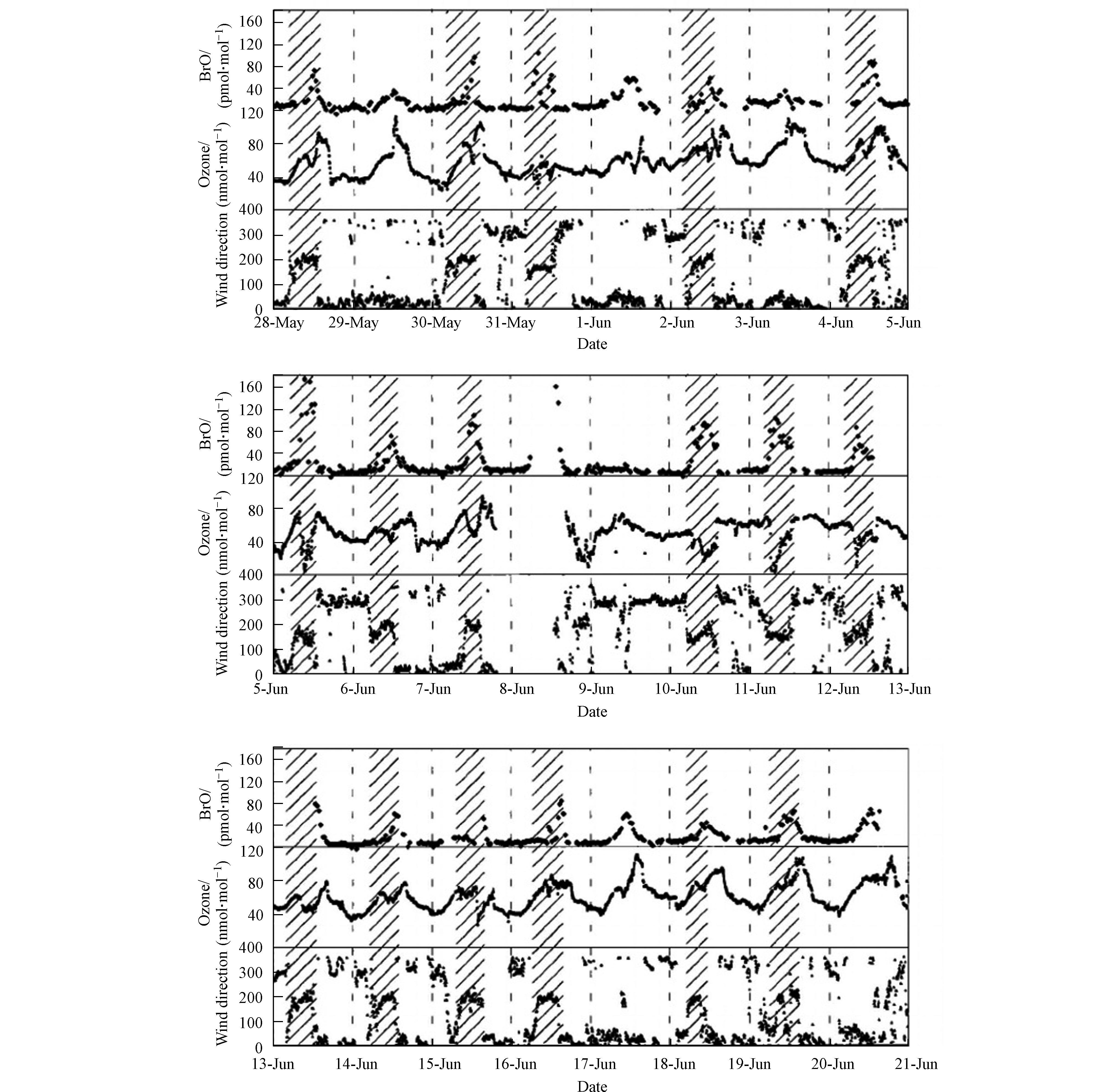
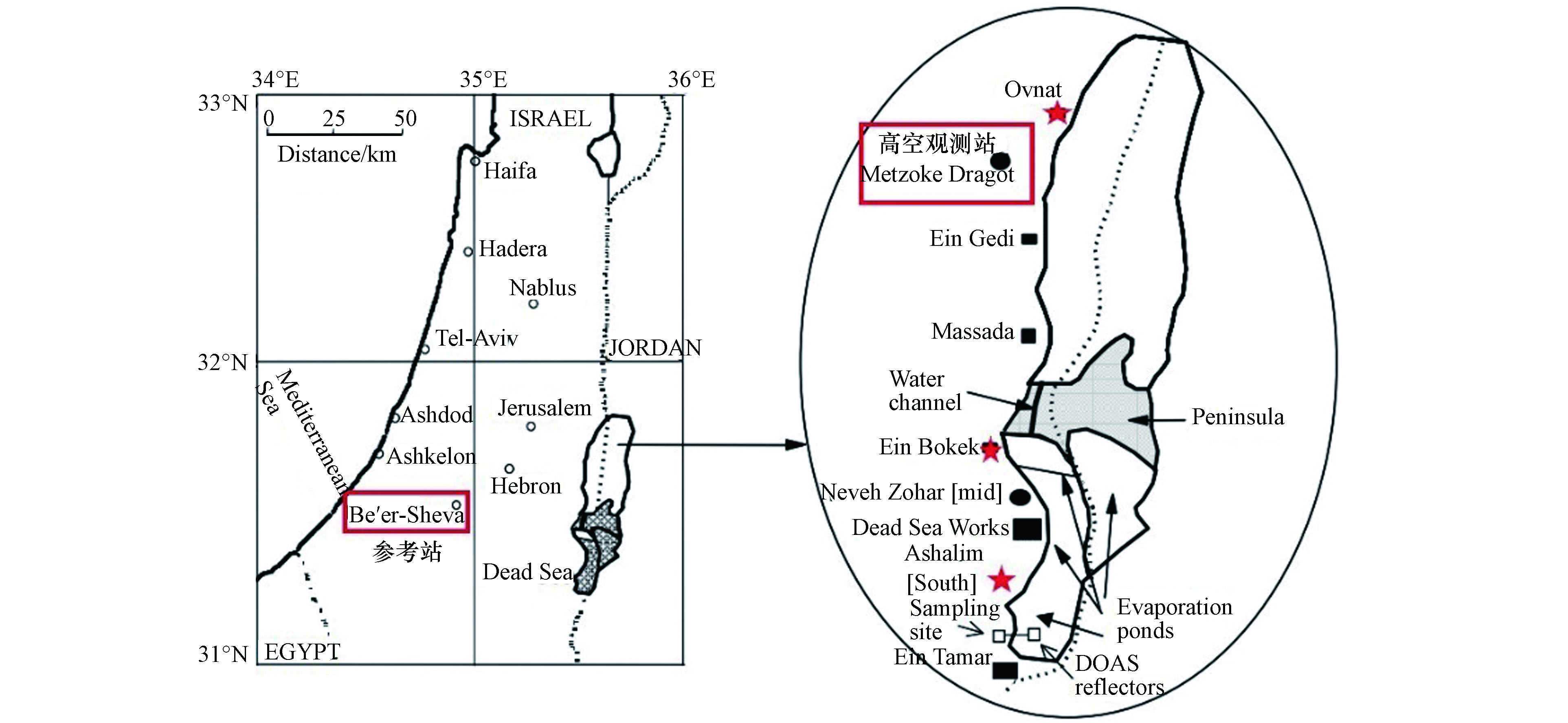
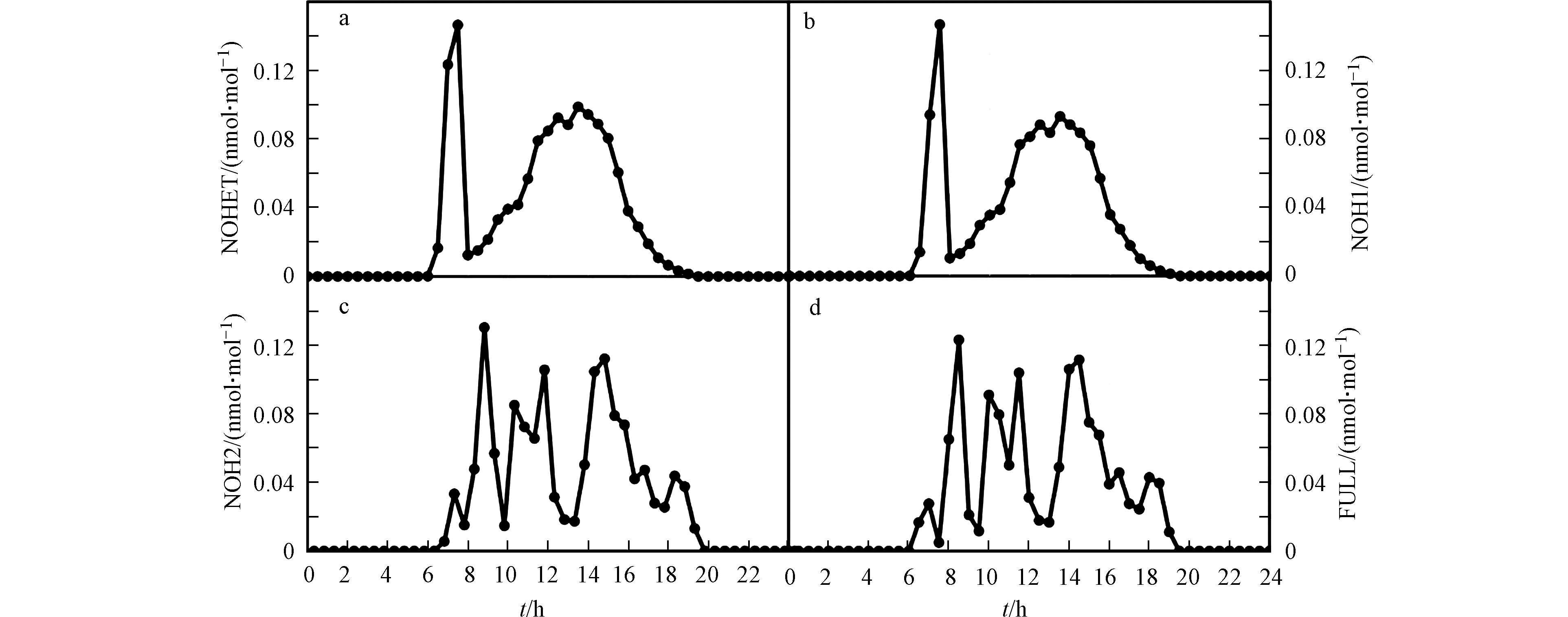
图 5 Tas等利用UAHCTM_1D模型模拟的在不同非均相反应开/关情况下BrO的日变化[36]a. NOHET-不含有非均相反应(R3)和(R4);b. NOH1-不含有非均相反应(R4);c. NOH2-不含有非均相反应(R3);d. FULL-含有完整的溴化学反应
Figure 5. Simulated BrO diurnal profiles in different scenarios using UAHCTM_1D model [36] a. NOHET-without heterogeneous reaction (R3) and (R4); b. NOH1-without heterogeneous reaction (R4): c. NOH2-without heterogeneous reaction (R3);d. FULL-with full bromine chemistry
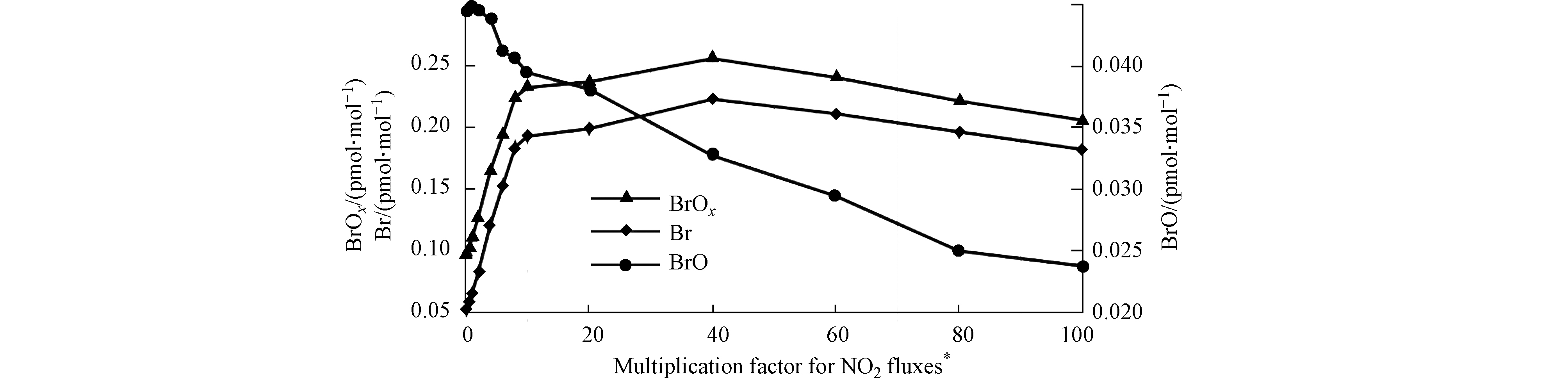
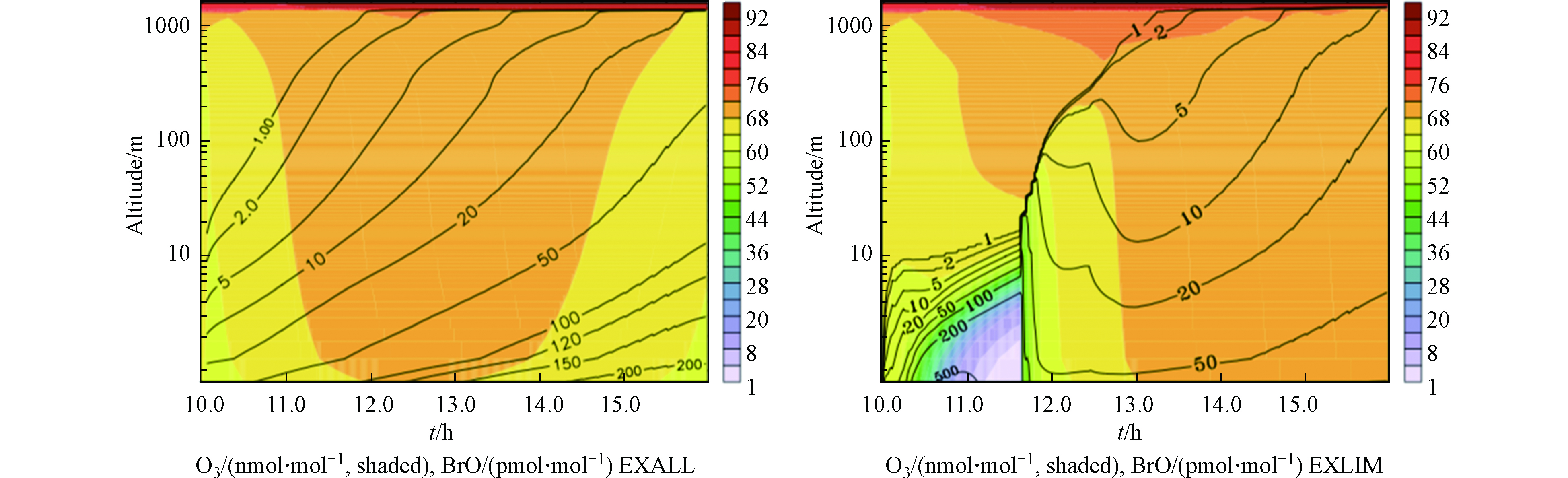
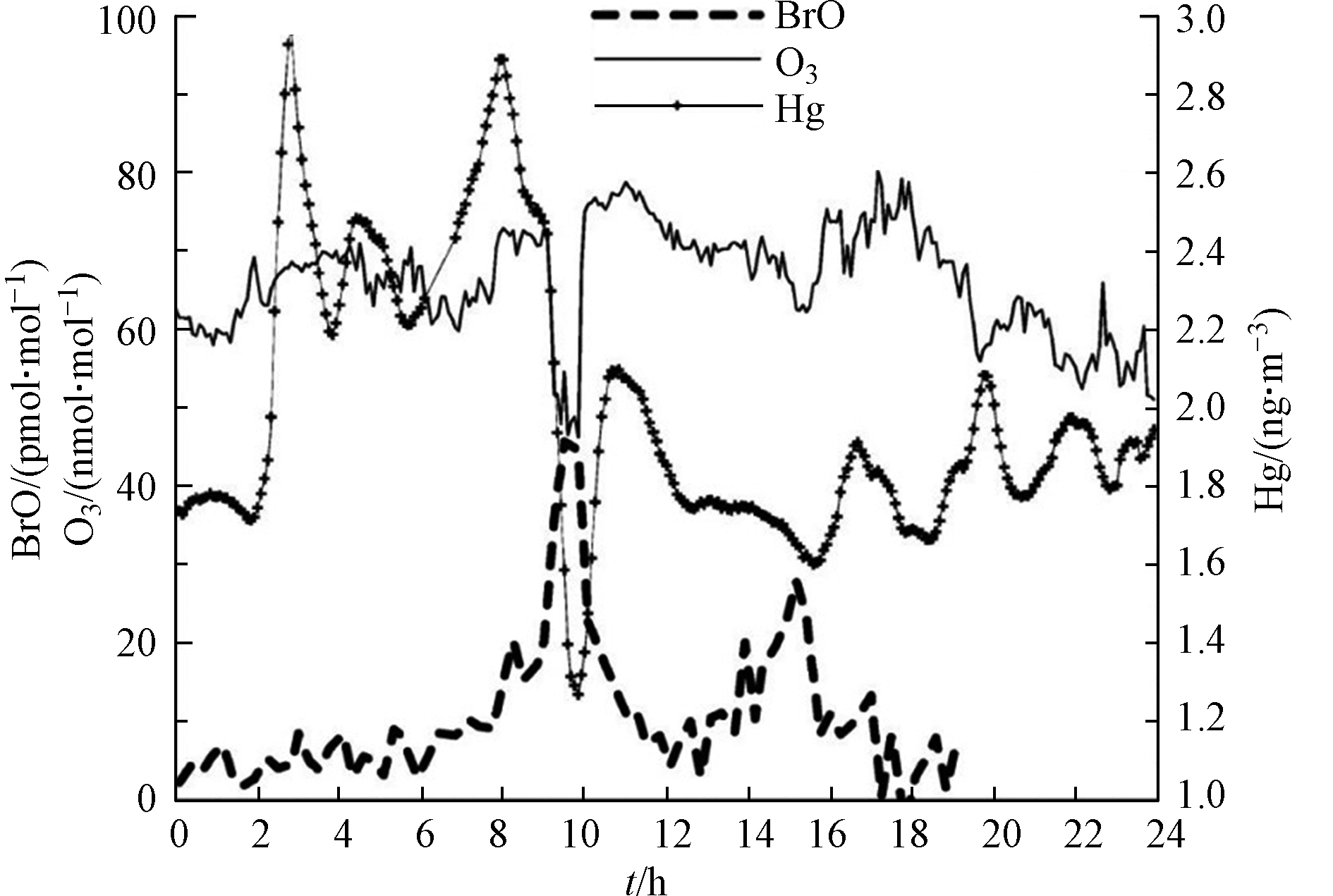
表 1 Tas等2001—2002年在死海周边的观测中各站点所处位置及观测条件 [2]
Table 1. Location and observation conditions of stations in the observation around the dead sea from 2001 to 2002 [2]
站点 Stations 观测条件 Observational conditions Ovant(死海北部) 死海西海岸,陆地观测 Ein Bokek(死海中部) 死海水体上空观测 Evaporation ponds DSW (死海南部) 蒸发盐池上空观测 Metzoke Dragot 高空(死海上方400m)及陆地观测 Ein Tamar 蒸发盐池南部,陆地观测  下载: 导出CSV
下载: 导出CSV
表 2 溴离子和氯离子在普通海水和死海海水中的浓度及其富集程度[2]
Table 2. Concentration and enrichment degree of bromine and chloride ions in ordinary seawater and dead sea seawater by Tas et al. [2]
站点 Station pH Cl−/(g·L−1) Br−/(g·L−1) Br−/Cl−(×103) 富集指数 ${\rm{Br}}_{{\rm{DS}}}^ - /{\rm{Br}}_{{\rm{OW}}}^ - $ 普通海水 19 0.065 3 死海北部(Qalya) 5.9 225 6.1 27 94 死海北部的中间
(Mitzpeh Shalem)5.7 255 6.9 27 106 死海中部(Ein Gedi) 5.8 236 6.4 27 98 蒸发盐池
(Ein Bokek)5.5 306 8.3 27 128 Neveh Zohar 5.0 354 9.6 27 148 死海南部 4.9 405 11.0 27 169
下载: 导出CSV
-
[1] NISSENBAUM A. Minor and trace elements in Dead Sea water [J]. Chemical Geology, 1977, 19(1/4): 99-111. [2] TAS E, PELEG M, MATVEEV V, et al. Frequency and extent of bromine oxide formation over the Dead Sea [J]. Journal of Geophysical Research Atmospheres , 2005, 110(D11): D11304. doi: 10.1029/2004JD005665 [3] HEBESTREIT K, STUTZ J, ROSEN D, et al. First DOAS measurements of tropospheric bromine oxide in mid latitudes [J]. Science, 1999, 283(5398): 55-57. doi: 10.1126/science.283.5398.55 [4] SIMPSON W R, von GLASOW R, RIEDEL K, et al. Halogens and their role in polar boundary-layer ozone depletion [J]. Atmospheric Chemistry and Physics, 2007, 7(16): 4375-4418. doi: 10.5194/acp-7-4375-2007 [5] MATVEEV V, PELEG M, ROSEN D,et al. Bromine oxide-ozone interaction over the Dead Sea [J]. Journal of Geophysical Research:Atmospheres, 2001, 106(D10): 10375-10387. doi: 10.1029/2000JD900611 [6] BARRIE L A. Arctic air pollution: An overview of current knowledge [J]. Atmospheric Environment, 1986, 20(4): 643-663. [7] OLTMANS S J, KOMHYR W D. Surface ozone distributions and variations from 1973-1984: Measurements at the NOAA Geophysical Monitoring for Climate Change Baseline Observatories [J]. Journal of Geophysical Research Atmospheres, 1986, 91(D4): 5229-5236. doi: 10.1029/JD091iD04p05229 [8] OLTMANS S J, SCHNELL R C, SHERIDAN P J, et al. Seasonal surface ozone and filterable bromine relationship in the high Arctic [J]. Atmospheric Environment, 1989, 23(11): 2431-2441. doi: 10.1016/0004-6981(89)90254-0 [9] BERG W W, SPERRY P D, RAHN K A, et al. Atmospheric bromine in the Arctic [J]. Journal of Geophysical Research:Atmospheres, 1983, 88(C11): 6719-6736. doi: 10.1029/JC088iC11p06719 [10] STURGES W T. Excess particulate and gaseous bromine at a remote coastal location [J]. Atmospheric Environment, 1990, 24(1): 167-171. doi: 10.1016/0960-1686(90)90452-S [11] BARRIE L A, BOTTENHEIM J W, SCHNELL R C, et al. Ozone destruction and photochemical reactions at polar sunrise in the lower Arctic atmosphere [J]. Nature, 1988, 334(6178): 138-141. doi: 10.1038/334138a0 [12] FAN S M, JACOB D J. Surface ozone depletion in Arctic spring sustained by bromine reactions on aerosols [J]. Nature, 1992, 359(6395): 522-524. doi: 10.1038/359522a0 [13] MCCONNELL J C, HENDERSON G S, BARRIE L, et al. Photochemical bromine production implicated in Arctic boundary-layer ozone depletion [J]. Nature, 1992, 355(6356): 150-152. doi: 10.1038/355150a0 [14] HAUSMANN M, PLATT U. Spectroscopic measurement of bromine oxide and ozone in the high Arctic during Polar Sunrise Experiment 1992 [J]. Journal of Geophysical Research Atmospheres, 1994, 99(D12): 25399-25413. doi: 10.1029/94JD01314 [15] BOTTENHEIM J W, BARRIE L A, ATLAS E, et al. Depletion of lower tropospheric ozone during Arctic spring: The Polar Sunrise Experiment 1988 [J]. Journal of Geophysical Research, 1990, 95(D11): 18555-18568. doi: 10.1029/JD095iD11p18555 [16] YOKOUCHI Y, AKIMOTO H, BARRIE L A, et al. Serial gas chromatographic/mass spectrometric measurements of some volatile organic-compounds in the Arctic atmosphere during the 1992 Polar Sunrise Experiment [J]. Journal of Geophysical Research:Atmospheres, 1994, 99(D12): 25379-25389. doi: 10.1029/94JD00227 [17] SIMPSON W R, KING M D, BEINE H J, et al. Atmospheric photolysis rate coefficients during the Polar Sunrise Experiment ALERT2000 [J]. Atmospheric Environment, 2002, 36(15/16): 2471-2480. [18] PLATT U , LEHRER E . ARCTOC final report (Arctic tropospheric ozone chemistry NO. EVSV-CT93-0318) [R]. European Community, 1997. [19] TANG T, MCCONNELL J C. Autocatalytic release of bromine from Arctic snow pack during polar sunrise [J]. Geophysical Research Letters, 1996, 23(19): 2633-2636. doi: 10.1029/96GL02572 [20] WENNBERG P. Bromine explosion [J]. Nature, 1999, 397(6717): 299-301. [21] HALFACRE J W, KNEPP T N, SHEPSON P B, et al. Temporal and spatial characteristics of ozone depletion events from measurements in the Arctic [J]. Atmospheric Chemistry and Physics, 2014, 14(10): 4875-4894. doi: 10.5194/acp-14-4875-2014 [22] MORIN S, HOENNINGER G, STAEBLER R M, et al. A high time resolution study of boundary layer ozone chemistry and dynamics over the Arctic Ocean near Alert, Nunavut [J]. Geophysical Research Letters, 2005, 32(8): L08809. [23] JONES A E, ANDERSON P S, WOLFF E W, et al. A role for newly forming sea ice in springtime polar tropospheric ozone loss? Observational evidence from Halley Station, Antarctica [J]. Journal of Geophysical Research Atmospheres, 2006, 111(D8): D08306. [24] JACOBI H W, KALESCHKE L, RICHTER A, et al. Observation of a fast ozone loss over frost flowers in the marginal ice zone of the Arctic Ocean [J]. Journal of Geophysical Research Atmospheres, 2006, 111(D15): D15309. doi: 10.1029/2005JD006715 [25] PLATT U. Differential optical absorption spectroscopy (DOAS), in air monitoring by spectroscopic techniques [J]. Chemical Analysis Series, 1994: 127. [26] TAS E, MATVEEV V, ZINGLER J, et al. Frequency and extent of ozone destruction episodes over the Dead Sea, Israel [J]. Atmospheric Environment, 2003, 37(34): 4769-4780. doi: 10.1016/j.atmosenv.2003.08.015 [27] SHECHNER M, GUENTHER A, RHEW R, et al. Emission of volatile halogenated organic compounds over various Dead Sea landscapes [J]. Atmospheric Chemistry and Physics, 2019, 19(11): 7667-7690. doi: 10.5194/acp-19-7667-2019 [28] HOLLA R, SCHMITT S, FRIEß U, et al. Vertical distribution of BrO in the boundary layer at the Dead Sea [J]. Environmental Chemistry, 2015, 12(4): 438-460. doi: 10.1071/EN14224 [29] SANDER R, KERKWEG A, JOCKEL P, et al. Technical note: The new comprehensive atmospheric chemistry module MECCA [J]. Atmospheric Chemistry and Physics, 2005, 5(2): 445-450. doi: 10.5194/acp-5-445-2005 [30] SANDER R, JOCKEL P, KIRNER O, et al. The photolysis module JVAL-14, compatible with the MESSy standard, and the JVal PreProcessor (JVPP) [J]. Geoscientific Model Development, 2014, 7(6): 2653-2662. doi: 10.5194/gmd-7-2653-2014 [31] SANDER R, BAUMGAERTNER A, GROMOV S, et al. The atmospheric chemistry box model CAABA/MECCA-3.0 [J]. Geoscientific Model Development, 2011, 4(2): 373-380. doi: 10.5194/gmd-4-373-2011 [32] BIAZAR A P . The role of natural nitrogen oxides in ozone production in the southeastern environment [D]. Huntsville : University of Alabama in Huntsville, 1995. [33] von GLASOW R, SANDER R, BOTT A, et al. Modeling halogen chemistry in the marine boundary layer 1. Cloud-free MBL [J]. Journal of Geophysical Research Atmospheres, 2002, 107(D17): 4341. [34] von GLASOW R, SANDER R, BOTT A, et al. Modeling halogen chemistry in the marine boundary layer 2. Interactions with sulfur and the cloud-covered MBL [J]. Journal of Geophysical Research Atmospheres, 2002, 107(D17): 4323. [35] VON GLASOW R . Modeling the gas and aqueous phase chemistry of the marine boundary layer [D]. Mainz, Germany: Johannes Gutenberg University of Mainz, 2001. [36] TAS E, PELEG M, PEDERSEN D U, et al. Measurement-based modeling of bromine chemistry in the boundary layer – 1: Bromine chemistry at the Dead Sea [J]. Atmospheric Chemistry and Physics, 2006, 6(12): 5589-5604. doi: 10.5194/acp-6-5589-2006 [37] TAS E, PELEG M, PEDERSEN D U, et al. Measurement-based modeling of bromine chemistry in the Dead Sea boundary layer - Part 2: The influence of NO2 on bromine chemistry at mid-latitude areas [J]. Atmospheric Chemistry and Physics, 2008, 8(16): 4811-4821. doi: 10.5194/acp-8-4811-2008 [38] SMOYDZIN L , VON GLASOW R. Modelling chemistry over the Dead Sea: bromine and ozone chemistry [J]. Atmospheric Chemistry and Physics, 2009, 9(14): 5057-5072. [39] TAS E, OBRIST D, PELEG M, et al. Measurement-based modelling of bromine-induced oxidation of mercury above the Dead Sea [J]. Atmospheric Chemistry and Physics, 2012, 12: 2429-2440. doi: 10.5194/acp-12-2429-2012 [40] WAYNE R P, POULET G, BIGGS P, et al. Halogen oxides: radicals, sources and reservoirs in the laboratory and in the atmosphere [J]. Atmospheric Environment, 1995, 29(20): 2677-2881. doi: 10.1016/1352-2310(95)90286-4 [41] HANSON D R, RAVISHANKARA A R. Heterogeneous chemistry of bromine species in sulfuric acid under stratospheric conditions [J]. Geophysical Research Letters, 1995, 22(4): 385-388. doi: 10.1029/94GL03379 [42] HANSON D R , RAVISHANKARA A R , LOVEJOY E R. Reaction of BrONO2 with H2O on submicron sulfuric acid aerosol and implications for the lower stratosphere [J]. Journal of Geophysical Research:Atmospheres, 1996, 101(D4): 9063-9069. [43] SANDER R, RUDICH Y, von GLASOW R, et al. The role of BrNO3 in marine tropospheric chemistry: A model study [J]. Geophysical Research Letters, 1999, 26(18): 2857-2860. doi: 10.1029/1999GL900478 [44] le BRAS G, PLATT U. A possible mechanism for combined chlorine and bromine catalyzed destruction of tropospheric ozone in the Arctic [J]. Geophysical Research Letters, 1995, 22(5): 599-602. doi: 10.1029/94GL03334 [45] SOLOMON S, GARCIA R R, RAVISHANKARA A R. On the role of iodine in ozone depletion [J]. Journal of Geophysical Research Atmospheres, 1994, 99(D10): 20491-20499. doi: 10.1029/94JD02028 [46] ADAMS J W, HOLMES N S, CROWLEY J N. Uptake and reaction of HOBr on frozen and dry salt surfaces between 253 and 233 K [J]. Atmospheric Chemistry and Physics, 2002, 2(1): 79-91. doi: 10.5194/acp-2-79-2002 [47] SAIZ-LOPEZ A, PLANE J M C, MAHAJAN A S, et al. On the vertical distribution of boundary layer halogens over coastal Antarctica: Implications for O3, HOx, NOx and the Hg lifetime [J]. Atmospheric Chemistry and Physics, 2008, 8(4): 887-900. doi: 10.5194/acp-8-887-2008 [48] AKIMOTO H . Atmospheric reaction chemistry [M]. Tokyo: Springer. 2016. [49] ZHOU J S, CAO L, LI S M. Influence of the background nitrogen oxides on the tropospheric ozone depletion events in the Arctic during springtime [J]. Atmosphere, 2020, 11(4): 344. doi: 10.3390/atmos11040344 [50] SCHROEDER W H, ANLAUF K G, BARRIE L A, et al. Arctic springtime depletion of mercury [J]. Nature, 1998, 394(6691): 331-332. doi: 10.1038/28530 [51] MOORE C W, OBRIST D, LURIA M, et al. Atmospheric mercury depletion events at the Dead Sea: Spatial and temporal aspects [J]. Atmospheric Environment, 2013, 69: 231-239. doi: 10.1016/j.atmosenv.2012.12.020 [52] OBRIST D, TAS E, PELEG M, et al. Bromine-induced oxidation of mercury in the mid-latitude atmosphere [J]. Nature Geoscience, 2011, 4(1): 22-26. doi: 10.1038/ngeo1018 [53] PELEG M, MATVEEV V, TAS E, et al. Mercury depletion events in the troposphere in mid-latitudes at the Dead Sea, Israel [J]. Environmental Science & Technology, 2007, 41(21): 7280-7285. [54] GRELL G A, PECKHAM S E, SCHMITZ R, et al. Fully coupled 'online' chemistry within the WRF model [J]. Atmospheric Environment, 2005, 39(37): 6957-6976. doi: 10.1016/j.atmosenv.2005.04.027 [55] BYUN D, SCHERE K L. Review of the governing equations, computational algorithms, and other components of the models-3 community multiscale air quality (CMAQ) modeling system [J]. Applied Mechanics Reviews, 2006, 59(2): 51-77. doi: 10.1115/1.2128636 [56] HÖRMANN C, SIHLER H, BEIRLE S, et al. Seasonal variation of tropospheric bromine monoxide over the Rann of Kutch salt marsh seen from space [J]. Atmospheric Chemistry and Physics, 2016, 16(20): 13015-13034. doi: 10.5194/acp-16-13015-2016 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 3523
- HTML全文浏览数: 3523
- PDF下载数: 110
- 施引文献: 0
