




-
我国是纺织印染大国,染料生产总量和贸易量位居世界第一。2015年全国工业废水的排放总量为199.5×109 t,其中染料行业排放废水18.4×109 t,约占全国工业排放废水的10%。蒽醌染料是仅次于偶氮染料的第2大染料,被广泛用作纤维素织物、羊毛和聚酰胺纤维的商业三色染色配方中的一次或二次染料,以增强染色效果[1]。溴氨酸钠(1-氨基-4-溴蒽醌-2-磺酸钠,Bromaminic acid sodium salt, BAA)是合成蒽醌类染料的重要染料中间体。BAA可溶于水,是工业废水中常见的污染物,属于不可生物降解、难降解的有机污染物[2]。由于染料生产工艺效率低下,部分染料在生产过程中未得到有效利用,最后直接以工业废水方式排放。大量BAA被排放到水体中,由于其具有生物毒性、结构稳定性和不可生物降解性等特点,对环境和健康造成严重危害。因此,寻找一种合适的处理BAA废水的方法迫在眉睫。
高级氧化法(advanced oxidation processes, AOPs)是一种有效降解水中难降解有机污染物的技术。在AOPs中,非均相催化臭氧氧化(Heterogeneous catalytic ozonation, HCO)因其氧化能力强、适用范围广、催化剂可回收性且不产生二次污染等多项优点而备受关注[3]。然而,催化剂的性质决定HCO的效率。为了提高HCO的效率,人们进行了大量的研究来开发新型催化剂。其中,MnO2因其价态多、结构多样、稳定性高、成本低等特点,被广泛用于催化臭氧化。到目前为止,为了实现高效的催化臭氧化,研究人员设计了不同的MnO2晶体形式,如α-、β-、γ-和δ-MnO2[4-6]。但二氧化锰晶体易团聚导致催化活性位点减少, 且会出现金属浸出,从而影响HCO的效率。硅藻土(diatomite, DE)具有高表面积、复杂的多孔结构、优异的耐热性等特点[7],同时,硅藻土天然孔隙结构和表面的羟基官能团可固定并吸附金属离子,降低金属溶解速度,减少二次污染。因此,将二氧化锰与硅藻土结合可以抑制金属浸出问题[8],防止超细金属氧化物纳米颗粒在溶液中团聚,从而显著提高HCO的效率。此外,硅藻土作为一种天然矿物,储量丰富、成本低、不存在二次污染[9]。
基于上述研究背景,本研究利用水热法将α-MnO2负载到DE表面,合成了α-MnO2/DE材料,利用系列表征手段对α-MnO2/DE的表面形貌和物相结构等进行了研究分析,且考察了α-MnO2/DE催化臭氧氧化降解BAA的性能,此外,分析了其活性氧(reactive oxygen species, ROS)生成和HCO的可能机理。最后,探究了α-MnO2/DE在实际应用中的降解效果。
-
高锰酸钾、一水合硫酸锰、过硫酸铵、硅藻土、溴氨酸钠、5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)、2,2,6,6-四甲基哌啶氧化物(TEMP)、对-苯醌、叔丁醇、糠醇、乙醇、硫酸、氢氧化钠、氯化钠、硝酸钠、硫酸钠、碳酸钠、磷酸钠、碳酸氢钠、铁氰化钾。实验用水为去离子水,以上所有化学品和试剂均为分析级,购自上海阿拉丁生物科技有限公司。
-
α-MnO2制备:将1.26 g KMnO4和0.51 g MnSO4∙H2O溶解于80 mL去离子水中,搅拌30 min。混合溶液转移反应釜中,然后将反应釜加热至160 ℃,升温速率为10 ℃ min−1,保温12 h。冷却至室温后,分别用去离子水和乙醇洗涤固体沉淀物5次,离心收集。粉末在80 ℃下干燥12 h。α-MnO2/DE制备:待KMnO4和MnSO4∙H2O充分溶解后,将1.21 g DE加入混合溶液,搅拌30 min,剩余步骤与制备α-MnO2相同。
β-MnO2制备:将MnSO4·H2O(2.71 g)和(NH4)2S2O8(3.65 g)混合在80 mL去离子水中。然后将混合物转移到反应釜中,在140 °C下加热12 h。分别用去离子水和乙醇洗涤固体沉淀物5次。最后,将固体在60 °C的空气中干燥12 h。β-MnO2/DE制备:待MnSO4·H2O和(NH4)2S2O8充分溶解后,将1.21 g DE加入混合溶液,搅拌30 min,剩余步骤与制备β-MnO2相同。
γ-MnO2制备:将MnSO4·H2O(3.38 g)和(NH4)2S2O8(4.58 g)混合在80 mL去离子水中。将混合溶液在室温下搅拌30min,然后将混合物转移到反应釜中,在90 ℃下加热24 h。分别用去离子水和乙醇洗涤固体沉淀物5次,在60 ℃空气中干燥12 h。γ-MnO2/DE制备:待MnSO4·H2O和(NH4)2S2O8充分溶解后,将1.21 g DE加入混合溶液,搅拌30 min,剩余步骤与制备γ-MnO2相同。
δ-MnO2制备:先将3 g KMnO4溶解50 mL去离子水中,然后加入20 mL 1.4 mol·L−1的葡萄糖水溶液。在室温下反应两小时后,得到棕色凝胶。凝胶在110 ℃下干燥24 h,在400 ℃下煅烧2 h。冷却至室温后,离心后用去离子水洗涤5次,收集固体沉淀物,去除残盐。粉末在80 ℃下干燥12 h[10]。δ-MnO2/DE制备:凝胶在110 ℃下干燥24 h后,将1.21 g DE与干燥后的粉末混合均匀,剩余步骤与制备δ-MnO2相同。
硅藻土在进行实验之前分别用去离子水与乙醇各洗涤5次,在烘箱中60 ℃干燥12 h,并将收集到的粉末命名为硅藻土(DE)。
-
以不同材料为催化剂,BAA为模型污染物,在有效容积为500 mL玻璃洗气瓶中对模拟废水进行非均相臭氧催化氧化降解实验。反应器中加入200 mL BAA,然后加入适量的催化剂,放置在25 ℃的水浴中,持续搅拌。用3S-T10型实验室臭氧发生器(同林科技,中国)将臭氧以500 mL·min−1的流速通过曝气头鼓泡到反应器底部,臭氧浓度通过调节电压来控制,用3S-J5000型台式臭氧浓度在线分析仪(同林科技,中国)测定臭氧浓度,随后,以预定的时间间隔抽出2 mL反应溶液,并通过0.45 µm尼龙过滤器过滤。所有实验重复3次,用误差棒表示平均值的标准差。
采用岛津紫外可见分光光度计(UV
2600 ,Shimadzu,Japan)在200~700 nm内扫描光谱,并在最大吸收峰(485 nm)处建立吸光度与BAA浓度的对应关系以检测BAA的浓度。采用ICP-MS(Agilent7500 , USA)测定金属离子浸出情况。用电子透射电镜(TEM; JEM-
1400 flash, Japan)和电子扫描电镜(SEM; Quanta 200 FEG, FEI Company, USA)观察样品的形貌和精细结构。采用X射线衍射(XRD; PANalytical X’Pert Pro, Netherlands)分析晶体结构。采用傅里叶变换红外光谱仪(VERTEX70)测量衰减全反射傅里叶变换红外光谱(FTIR)。采用比表面积测试法(BET; Autosorb - IQ3, USA)测定样品的BET比表面积。采用循环伏安法(CV)、电化学阻抗法(EIS)(三电极系统电化学工作站; CHI760e,上海陈华仪器有限公司)对催化剂电化学性能进行表征。利用X射线光电子能谱(XPS; Thermo, USA)测定元素组成。使用电子自旋共振(ESR/EPR,JEOL JES-FA200 ESR)鉴定活性氧物种。 -
利用XRD对不同材料的晶体结构进行了研究,结果如图1(a)所示。可见, α-、β-、γ-和δ-MnO2的衍射峰分别与JCPDS29-
1020 、JCPDS24-0735、JCPDS14-0644和JCPDS80-1098 的衍射峰匹配良好,与姚宏嘉等的研究[11]结果一致,验证了不同晶相MnO2的成功制备。如图1(b)所示,四种二氧化锰掺杂DE后合成的α-MnO2/DE、β-MnO2/DE、γ-MnO2/DE和δ-MnO2/DE与对应晶相的二氧化锰的衍射峰匹配良好[12]。此外,在2θ=26.6°附近的衍射峰对应于硅藻土SiO2的(101)晶面(JCPDS:46-1045 )。4种复合纳米材料的衍射图谱集合了MnO2和硅藻土的特征衍射峰,表明本研究已成功合成α-MnO2/DE、β-MnO2/DE、γ-MnO2/DE和δ-MnO2/DE。利用SEM观察各材料的微观形貌。由图2(a)~(d)所示,4种晶相的二氧化锰呈现出不同的形态,包括针状(α-MnO2)、棒状(β-MnO2)、海胆状(γ-MnO2)和球状(δ-MnO2)形貌。由图2(e)可见,呈圆盘状的硅藻土表面上分布着大量的孔洞结构。这些孔洞可为MnO2的负载提供大量的位点,有利于MnO2的负载和分散。由图2(f)~(i)可见,α-、β-、γ-和δ-MnO2均匀分布在硅藻土表面。此外,负载在DE上的MnO2分散性更好且粒径更小。因此,硅藻土作为载体,能有效改善MnO2的分散程度,使MnO2暴露出更多的活性位点,从而有利于提高材料的催化活性。
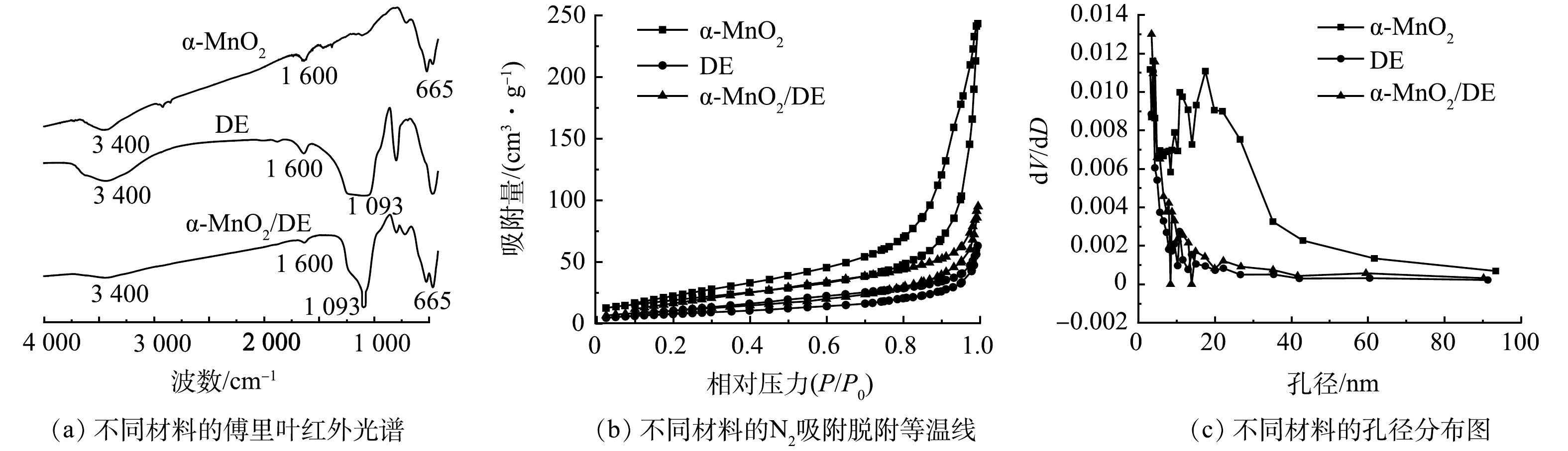
在α-MnO2/DE的HRTEM图像(图3(a))中,硅藻土保持了完整的多孔结构,可以清楚地观察到线状的α-MnO2均匀地分散在硅藻土的表面和孔径内,硅藻土的天然多孔结构得到了有效利用。此外,在α-MnO2/DE中可以观察到晶格间距为0.69 nm的点阵条纹(图3(b)),这与XRD中α-MnO2(JCPDS29-
1020 )(110)晶面的间距一致[13]。α-MnO2/DE的元素映射结果(图3(c)~(f))表明,Mn、O、Si元素均匀地分布在硅藻土的表面和孔洞内,再次证实了α-MnO2/DE复合材料的成功制备。图4(a)为DE、α-MnO2和α-MnO2/DE复合材料的红外光谱图。α-MnO2/DE中665 cm−1的峰可被分配给Mn—O的弯曲振动[14]。而1 093.57 cm−1处的峰是由硅藻土中Si—O—Si键的不对称伸缩振动造成的[15]。此外,约1 600 cm−1的波段被分配给表面羟基或水在拉伸模式下的特征峰[16],约3 400 cm−1处的峰值归因于表面羟基的弯曲振动[17]。
不同材料的吸附曲线和孔径分布如图4(b)~(c)所示。DE、α-MnO2和α-MnO2/DE表现出典型的IV等温线和H3型滞后环,显示出介孔结构[18]。表1总结了3种材料的比表面积和孔径分布。DE、α-MnO2和α-MnO2/DE的平均孔径分别为3.81、3.83、和3.43 nm,进一步证实了2种催化剂均为介孔材料。α-MnO2/DE的比表面积为41.23 m2·g−1,远高于硅藻土(28.13 m2·g−1),与未负载催化剂的硅藻土相比,α-MnO2/DE拥有更大的比表面积,这使得α-MnO2/DE具有更好的吸附性和催化活性。
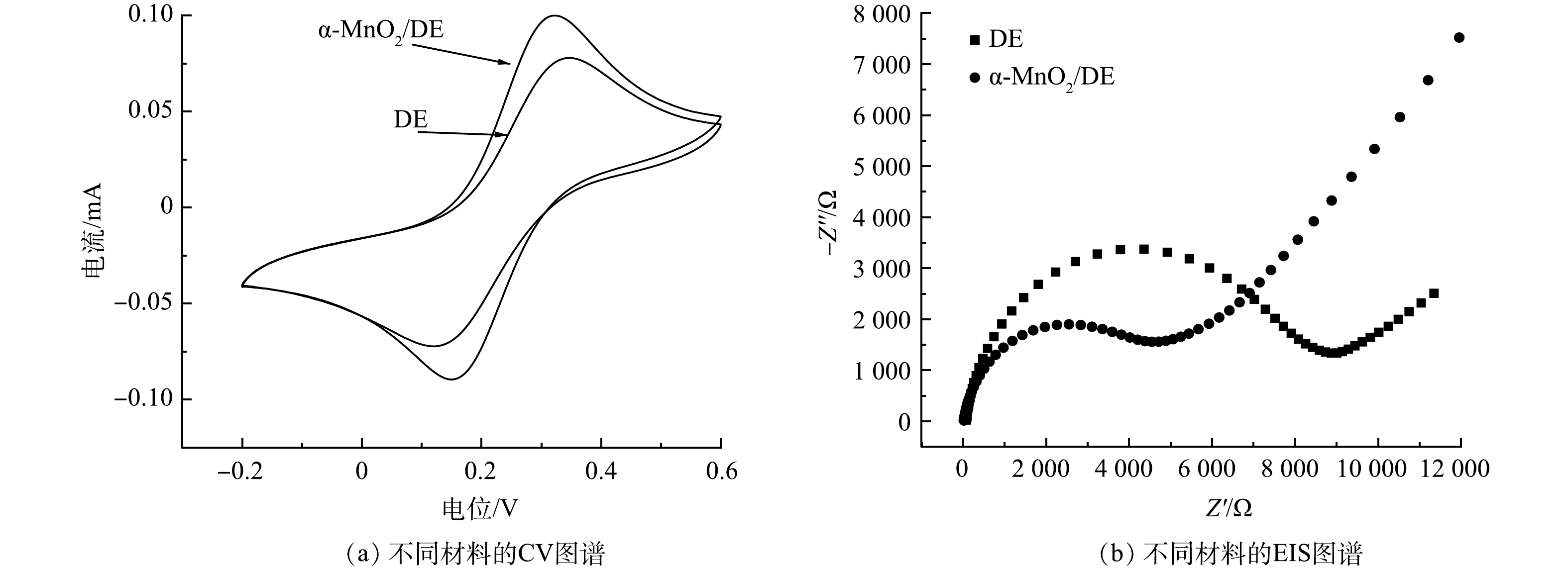
循环伏安曲线(CV)可表达材料的电子可转移性。由图5(a)可见,α-MnO2/DE的氧化峰电流比硅藻土高0.02 mA,表明α-MnO2/DE的电子转移效率更高,具有比硅藻土更好的氧化还原性能。此外,采用电化学阻抗谱(EIS)测试分析了DE和α-MnO2/DE的电子传递速率。理论上,半圆直径代表极化电阻的强度[19],电极电位的增大使得半圆直径增大,这意味着电荷转移电阻增大[20]。如图5(b)所示,α-MnO2/DE的奈奎斯特弧半径较小,表明α-MnO2/DE的极化电阻较低,电荷转移速率较快。这些结果证实了α-MnO2/DE加速了界面电子转移,有利于催化反应过程中性能的提升。
-
考察了不同催化剂、催化剂投加量、溶液pH、温度、臭氧浓度、初始BAA浓度等因素对臭氧催化氧化降解BAA效率的影响。实验条件为:50 mg·L−1 BAA,0.75 g·L−1催化剂,O3质量浓度5 mg·L−1,溶液pH为7,温度25 ℃。
不同晶相的MnO2对BAA吸附效果如图6(a)所示。α-MnO2对BAA的吸附率为10.21%,而其他3种MnO2对BAA的吸附率均小于10%。如图6(b)所示,将MnO2负载到硅藻土上后各复合材料对BAA的吸附率均得到提升,其中α-MnO2/DE对BAA的吸附率最高,达到17.87%,这得益于α-MnO2更高的比表面积和孔隙体积[21]。如图6(c)所示,单独O3在40 min对BAA的去除率为35.62%,而在O3和催化剂同时存在的情况下,BAA的在40 min的去除率分别达到52.72%(α-MnO2/O3)、48.26%(β-MnO2/O3)、52.58%(γ-MnO2/O3)和41.64%(δ-MnO2/O3),不同晶相MnO2对BAA的降解顺序为α-MnO2>γ-MnO2>β-MnO2>δ-MnO2,这得益于α-MnO2隧道结构能提供更多的活性位点[22]。将MnO2负载到硅藻土上后各复合纳米材料对BAA的去除率均得到显著提升,如图6(d)所示,α-MnO2/DE、β-MnO2/DE、γ-MnO2/DE和δ-MnO2/DE对BAA的降解率分别为76.27%、57.94%、65.77%和50.41%。通过对上述不同材料的对比可以发现,这些材料均能够在不同程度催化臭氧氧化降解BAA,而不同晶相的二氧化锰中α-MnO2对BAA的降解率最高,并且掺杂DE后对BAA的去除率提升效果最明显,提升了23.55%。将α-MnO2负载到硅藻土改善了α-MnO2的团聚程度,增加了更多的活性位点,因而使得催化剂的催化效率和催化活性均有一定的提高。因此,本实验选定α-MnO2/DE进行后续的实验研究。
α-MnO2/DE用量对BAA降解效果的影响情况如图7(a)所示。可见,随着催化剂含量从0 g·L−1增加到0.5 g·L−1,BAA的降解率在40 min内由35.62%稳步提高到81.39%。然而,进一步增加α-MnO2/DE的投加量至0.75 g·L−1 BAA的降解率没有得到显著提高,仅上升了1.02%。这是由于溶液中O3的浓度是固定的,因此,过量的催化剂会导致ROS瞬间过量形成。过量的ROS会与O3、自身或BAA中间体发生反应,可以较大程度上降低其与BAA发生反应的概率[23]。因此,在臭氧催化氧化过程中,0.5 g·L−1的α-MnO2/DE被确定为最佳用量,过量的催化剂用量不仅会增加运行成本,还会影响降解效率。
溶液pH能够通过调节催化剂的表面电荷、目标污染物的质子化、臭氧的分解动力学等对催化臭氧化过程产生显著影响。如图7(b)所示,当初始pH从3增加到11时,催化臭氧化对BAA的降解均有所提高,并在pH=11时达到了最高的降解率(91.27%)。这是因为在碱性条件下,BAA主要以去质子形式存在,并与O3快速反应[24],因而碱性条件能显著提升α-MnO2/DE/O3体系对BAA的降解率。
图7(c)为不同反应温度下,α-MnO2/DE臭氧催化氧化BAA的降解情况。将温度从5 ℃提高到25 ℃,BAA的降解效率在40 min内从72.15%提高到92.35%。这是由于过低的温度会降低O3在溶液中的溶解度,从而影响BAA的降解率[25]。而35 ℃下BAA的降解效率略微下降到84.9%。因此,α-MnO2/DE作为臭氧氧化催化剂具有较宽的反应温度适应范围。
O3剂量对O3/α-MnO2/DE降解BAA的影响如图7(d)所示。当O3投加量为2.5 mg·L−1时,在20 min时BAA的降解率为61.6%,增加臭氧剂量至5 mg·L−1,BAA的降解效率明显提高(92.35%)。进一步增加O3剂量至7.5 mg·L−1和10 mg·L−1时,对BAA的降解效果的提升可以忽略不计。这是由于催化剂的量是一定的,其体系内的活性位点数量也是一定的,过量的O3无法与催化剂上的活性位点充分接触产生自由基,进而对BAA的降解效率的提升也有限。考虑到运行成本,在我们的系统中,5 mg·L−1 O3对于BAA降解是足够的剂量。此外,考察了不同BAA浓度对α-MnO2/DE催化效率的影响,结果如图7(e)所示,在BAA质量浓度为50 mg·L−1,该体系在30 min内即可完全降解BAA,随着BAA浓度的增加,产生的中间产物更多,这些中间产物可竞争自由基,占据了部分活性位点,消耗了更多的自由基,使降解效率降低[26]。
通过以上实验,确定最佳实验条件为:BAA质量浓度50 mg·L−1,臭氧质量浓度5 mg·L−1, α-MnO2/DE投加量0.5 g·L−1,pH=11,温度25 ℃时,30 min内BAA降解率可达100%。
-
1) i-t/XPS分析。在臭氧催化氧化过程中对α-MnO2和α-MnO2/DE进行了计时电流法测试,结果如图8(a)所示。测量了先加入BAA再加入O3时电流的变化。对于α-MnO2和α-MnO2/DE,先加入BAA都没有引起电流的明显变化,说明BAA的吸附可以忽略不计[27]。随后O3的引入导致两种催化剂的电流波动明显,但α-MnO2/DE的电流波动更明显。这些观察结果证明了O3在α-MnO2/DE上的分解比在α-MnO2/DE上的分解更快。O3可以吸附在催化剂表面,通过电子转移反应生成ROS。考虑到α-MnO2也表现出明显的O3吸附和活化能力,我们有理由认为催化剂表面的Mn位点起到了活性位点的作用,主导了O3的分解和ROS的生成。
为了进一步证明α-MnO2/DE作为催化剂的反应活性位点,我们明确对比了新鲜和使用过的α-MnO2/DE的XPS表征分析。如图8(b)~(c)所示,α-MnO2/DE反应后的Mn2p3/2光谱可分为位于640.6、641.6和642.9 eV的3个峰,分别对应Mn2+、Mn3+和Mn4+的结合能[6]。相较于反应前,Mn3+的含量由63.7%下降到40.6%,Mn4+含量由36.3%上升到39.1%,Mn2+含量由0上升到20.3%,表明活性位点是富电子的Mn(Ⅲ),其可向臭氧提供电子并被氧化成Mn(Ⅳ)。Mn(Ⅲ)转化为Mn(Ⅳ)可以有效地诱导O3分解产生活性氧降解污染物,而降解污染物提供的电子也可以有效地促进Mn(Ⅳ)转化为Mn(Ⅲ)。
2)活性氧物种的确定及分析。为了确定反应过程中产生的ROS及其对BAA降解的贡献,使用ROS淬灭剂,如叔丁醇(消除·OH)[28],对-苯醌(消除·O2−)[29]和糠醇(消除1O2)[30]对体系中可能产生的自由基进行了推测,结果如图9(a)所示。随着对-苯醌和糠醇的加入,BAA的降解效果受到轻微抑制,说明·O2−和1O2可能不是导致BAA降解的主要ROS。与此相反,在叔丁醇存在时,BAA的降解效果受到了明显的抑制,降解率下降到37.76%,表明·OH在BAA的降解过程中起到了主要作用。
利用电子顺磁共振(ESR)进一步验证了·OH、·O2−和1O2在催化臭氧化过程中的作用。由图9(b)可见,DMPO-·OH加合物在单独O3和O3/α-MnO2/DE体系中均观察到强度比为1:2:2:1的特征光谱[31]。在单独O3和O3/α-MnO2/DE体系中也均观察到强度比1:1:1:1 DMPO-·O2−加合物(图9(c))的特征光谱与强度比为1:1:1的TEMP-1O2加合物(图9(d))的特征光谱[32]。此外,α-MnO2/DE的加入显著提高了DMPO-·OH的强度,使体系内产生了更多的·OH。然而,α-MnO2/DE的加入并没有显著影响DMPO-·O2−和TEMP-1O2的信号[33]。上述结果再次表明,在α-MnO2/DE催化臭氧氧化降解BAA的过程中,·OH为主要的ROS。
3)降解机理分析。基于以上分析结果,提出了α-MnO2/DE在臭氧催化氧化体系中可能的降解机理:首先,O3吸附在α-MnO2/DE催化剂表面富电子的活性位点Mn(Ⅲ)上且将其氧化成Mn(Ⅳ)。同时,O3被激活生成了ROS,并通过链式反应迅速生成·OH。·OH具有很高的反应活性,能够攻击和破坏BAA的分子结构,导致BAA最终矿化为CO2、H2O和无机离子。此外,降解污染物提供的电子也可以重新供应给富电子的Mn位点,有效促进Mn(Ⅳ)通过自由基反应链还原成Mn(Ⅲ)。综上所述,推测 α-MnO2/DE催化臭氧氧化产生·OH降解BAA的可能过程如式(1)~式(6)所示。
-
水和废水中存在多种类型的无机阴离子,它们会竞争体系内的·OH从而抑制臭氧化过程中污染物的降解[34, 35]。将几种常见阴离子(CO32−、HCO3−、Cl−、PO43−、NO3−和SO42−,各10 mmol·L−1)分别加入到α-MnO2/DE催化臭氧氧化BAA反应中,观察了BAA的降解情况(图10(a))。结果表明,阴离子对BAA的催化降解仅有轻微的抑制作用,其抑制作用顺序为Cl−>NO3−>PO43−>HCO3−>SO42−>CO32−。使降解率分别降低了16.72%、7.18%、6.14%、4.88%,2.2%和0.95%。Cl−是一种·OH清除剂,加入Cl−会淬灭反应体系中的·OH,进而可能产生较强的降解抑制作用。NO3−等阴离子可能会吸附在催化剂表面造成催化剂中毒,使催化活性下降。CO32−对BAA的去除效果没有显著影响可能归因于其对反应过程中pH的变化较小[36]。
为了进一步检验α-MnO2/DE的实际应用性能,考察了不同水基质:去离子水、市政供水、斛兵塘水(城市内湖)和染料359废水对BAA去除效果的影响,实验条件同2.2优化后的最佳实验条件。如图10(b)所示,其中染料359废水与纯水相比,BAA的去除率略有下降(71.57%),这可能归因于染料废水的复杂性,各种污染物优先被氧化为其余物质导致水样中BAA的降解率降低。而市政供水、斛兵塘水中BAA的降解率分别为86.72%和84.39%,与去离子水中BAA的降解率(92.35%)接近,对系统几乎无影响。这些结果表明,α-MnO2/DE系统用于实际废水处理是可行的。
此外,研究了α-MnO2/DE的可重复使用性和稳定性。如图10(c~d)所示,在5次循环利用后,α-MnO2/DE仍能有效降解83.65%的BAA。此外,α-MnO2/DE的锰离子浸出质量浓度较低,仅为0.26 mg·L−1,这归因于硅藻土表面的羟基官能团可固定并吸附金属离子,从而使锰离子与配体之间形成良好的结合。以上实验结果表明α-MnO2/DE具有优异的稳定性。
-
1)利用水热法成功制备出α-MnO2/DE复合纳米材料,α-MnO2/DE改善了α-MnO2的团聚程度,其更低的极化电阻以及更快地电子转移效率均有利于提高材料的催化性能。
2)在最佳实验条件(BAA质量浓度50 mg·L−1,臭氧质量浓度5 mg·L−1,催化剂投加量0.5 g·L−1,pH=11,温度25 ℃)下,α-MnO2/DE/O3体系在30 min内对BAA降解率可达100%。
3) Mn(Ⅲ)作为催化剂活性位点吸附O3并活化产生 ROS。其中,·OH作为α-MnO2/DE/O3体系内主要的ROS,能够有效降解BAA。
4) α-MnO2/DE具有较强的抗干扰性和实际使用性。在多种阴离子、不同水基质以及较宽的pH和温度范围内都表现出高效的催化去除有机污染物的能力。此外,硅藻土能够限制催化剂中金属离子的浸出,提升催化剂的稳定性和重复使用性。
硅藻土负载α-MnO2纳米复合材料催化臭氧氧化溴氨酸钠的性能及机理
Performance and mechanism of diatomite-loaded α-MnO2 nanocomposites for catalytic ozone oxidation of bromamine acid sodium salt
-
摘要: 采用水热法成功合成了4种晶相结构的MnO2及将其负载在硅藻土上的纳米复合材料,并对其催化臭氧氧化降解溴氨酸钠(BAA)性能进行了详细研究。采用XRD、SEM、CV、EIS等手段对材料进行表征,α-MnO2/DE更低的极化电阻以及更快地电子转移效率是其性能优异的原因。单因素实验结果表明,α-MnO2/DE催化臭氧氧化反应体系能在30 min内将50 mg·L−1 BAA完全降解。通过对反应前后XPS表征、自由基淬灭实验、ESR分析研究该反应体系的催化机理。结果表明,Mn(Ⅲ)作为反应活性位点使O3吸附并活化产生ROS,同时该体系的主要活性氧物种为·OH。此外,离子干扰实验和循环实验证明该新型催化剂具有优异的稳定性和广阔的应用前景。Abstract: MnO2 with four types of crystalline phase structure and the composite nanomaterials of these MnO2 loaded on diatomite were successfully synthesized by a hydrothermal method, and a detailed research was conducted on their performance in catalytic ozonation and degradation of bromamine acid sodium salt (BAA). The materials were characterized by XRD, SEM, CV and EIS, of which α-MnO2/DE presented the lower polarization resistance and the faster electron transfer efficiency, which contributed to its excellent performance. Single factor experiments showed that the α-MnO2/DE-catalyzed ozone oxidation system could degrade 50 mg·L−1 BAA by 100% within 30 min. XPS characterization before and after the reaction, free radical quenching experiment, and ESR analysis was used to identify the catalytic mechanism of the system. The results showed that Mn(Ⅲ) acted as the reactive site to adsorb O3 molecules and activate them to produce ROS, at the same time the main reactive oxygen species in the system was ·OH. In addition, ionic interference and cycling experiments demonstrated that the novel catalyst had an excellent stability and promising application for organic pollutant degradation.
-
Key words:
- heterogeneous catalytic ozonation /
- MnO2 /
- diatomite /
- bromamine acid sodium salt /
- mechanism.
-

-
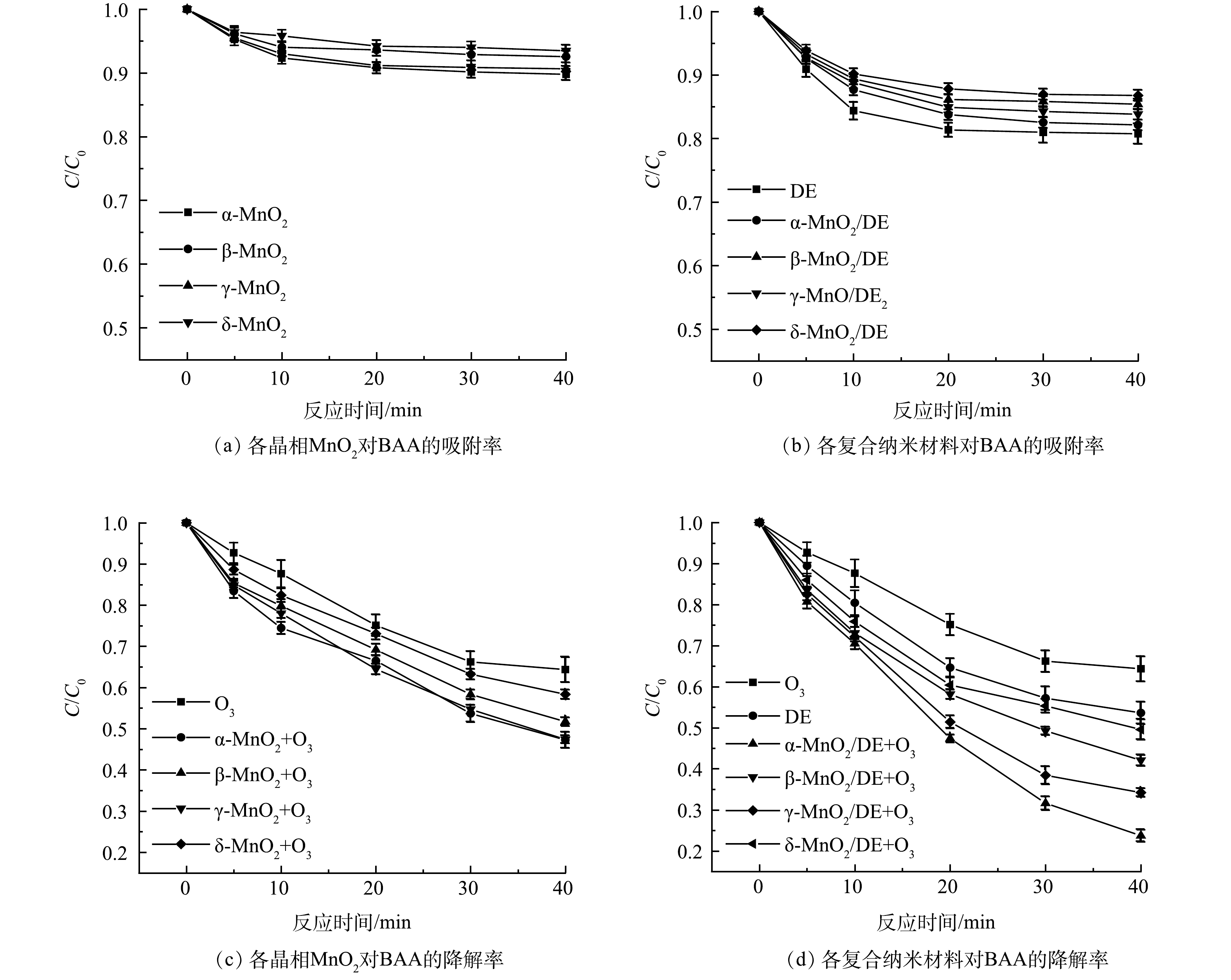
图 6 各材料对BAA的吸附率和各材料对BAA的降解率
Figure 6. Adsorption rate and degradation rate of BAA by each material

图 7 不同实验条件对BAA降解率的影响
Figure 7. Effect of different experimental conditions on the degradation rate of BAA
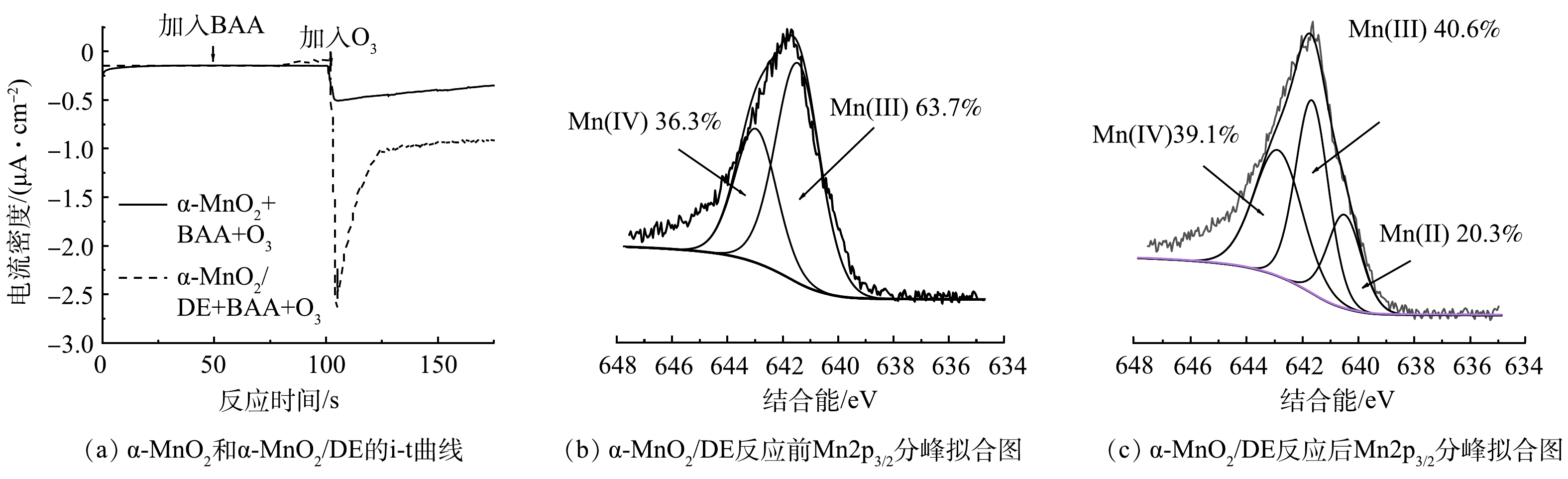
图 8 α-MnO2和α-MnO2/DE的i-t曲线与反应前后 α-MnO2/DE的Mn2p3/2 XPS 曲线分析
Figure 8. The i-t curves of α-MnO2 and α-MnO2/DE and Mn2p3/2 XPS spectra of α-MnO2/DE before and after the reaction
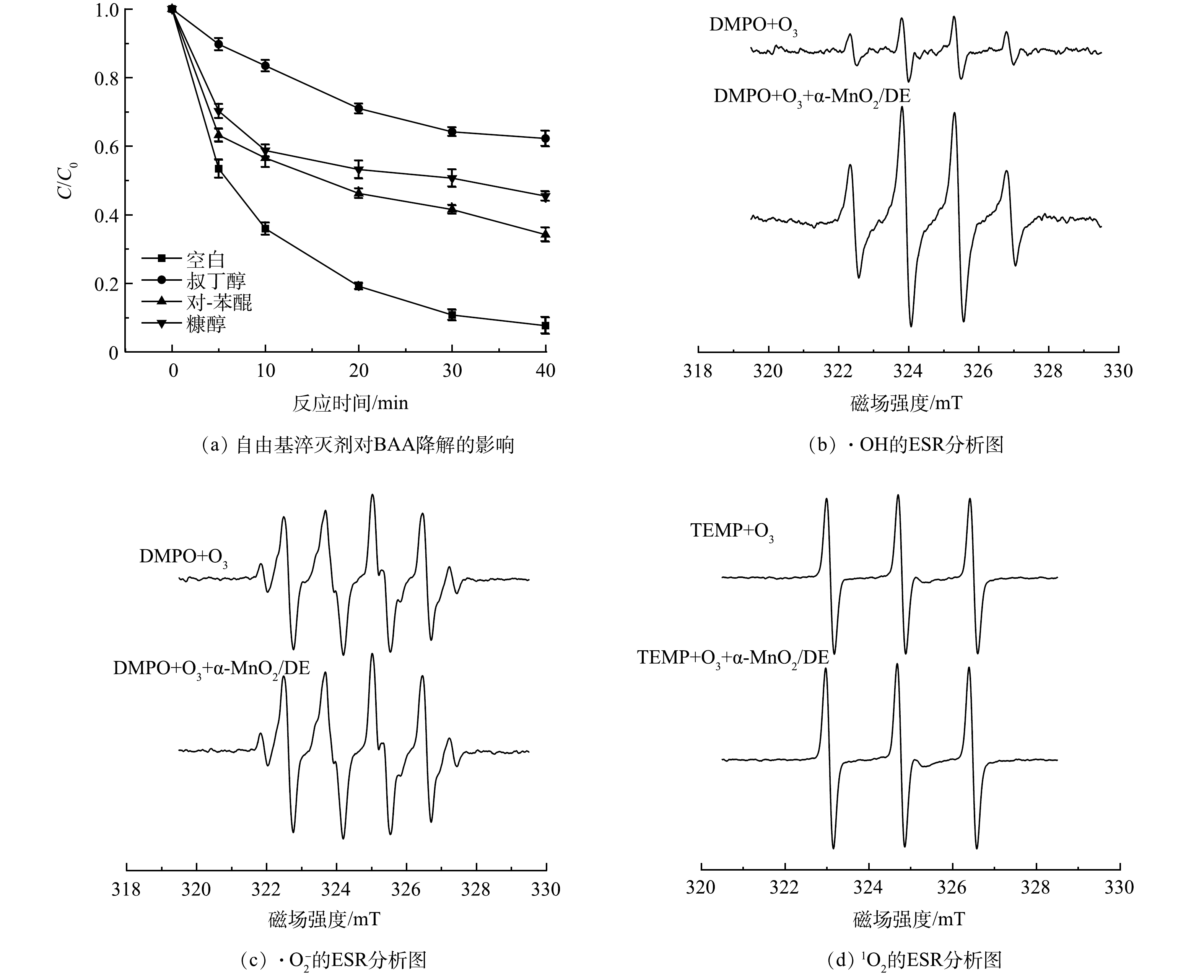
图 9 自由基淬灭剂对BAA降解的影响和α-MnO2/DE的ESR谱图
Figure 9. Effect of radical scavengers on degradation of BAA and ESR spectra of α-MnO2/DE
表 1 不同催化剂的结构特性
Table 1. Structural properties of the catalysts
样品 比表面积/(m2·g−1) 总孔容/(cm3·g−1) 平均孔径/nm DE 28.13 0.09 3.81 α-MnO2 88.61 0.37 3.83 α-MnO2/DE 41.23 0.13 3.41  下载: 导出CSV
下载: 导出CSV
-
[1] 邱沪生, 蔡天明, 陈立伟. 催化湿式过氧化法处理蒽醌-2-磺酸钠废水[J]. 环境工程学报, 2014, 8(4): 1497-502. [2] ROUTOULA E, PATWARDHAN S V. Degradation of anthraquinone dyes from effluents: A review focusing on enzymatic dye degradation with industrial potential[J]. Environmental Science & Technology, 2020, 54(2): 647-664. [3] YUAN Y, XING G, GARG S, et al. Mechanistic insights into the catalytic ozonation process using iron oxide-impregnated activated carbon[J]. Water Research, 2020, 177: 115785. doi: 10.1016/j.watres.2020.115785 [4] HUANG K Z, ZHANG H. Direct electron-transfer-based peroxymonosulfate activation by iron-doped manganese oxide (δ-MnO2) and the development of galvanic oxidation processes (GOPs)[J]. Environmental Science & Technology, 2019, 53(21): 12610-12620. [5] NAWAZ F, CAO H, XIE Y, et al. Selection of active phase of MnO2 for catalytic ozonation of 4-nitrophenol[J]. Chemosphere, 2017, 168: 1457-1466. doi: 10.1016/j.chemosphere.2016.11.138 [6] WANG F, DAI H, DENG J, et al. Manganese oxides with rod-, wire-, tube-, and flower-like morphologies: Highly effective catalysts for the removal of toluene[J]. Environmental Science & Technology, 2012, 46(7): 4034-4041. [7] DEHESTANIATHAR S, KHAJELAKZAY M, RAMEZANI-FARANI M, et al. Modified diatomite-supported CuO–TiO2 composite: Preparation, characterization and catalytic co oxidation[J]. Journal of the Taiwan Institute of Chemical Engineers, 2016, 58: 252-258. doi: 10.1016/j.jtice.2015.05.030 [8] TAN Y, LI C, SUN Z, et al. Natural diatomite mediated spherically monodispersed CoFe2O4 nanoparticles for efficient catalytic oxidation of bisphenol a through activating peroxymonosulfate[J]. Chemical Engineering Journal, 2020, 388: 124386. doi: 10.1016/j.cej.2020.124386 [9] 韩琳, 陈宋辉, 于鹏, 等. 磁性硅藻土的制备及其性能[J]. 环境工程学报, 2016, 10(6): 2987-2991. doi: 10.12030/j.cjee.201501046 [10] XIAO X, ZHANG Z, WU Y, et al. Ultrahigh‐loading manganese‐based electrodes for aqueous batteries via polymorph tuning[J]. Advanced Materials, 2023, 35(33): 2211555. doi: 10.1002/adma.202211555 [11] 姚宏嘉, 陈星, 张玉, 等. 生物炭负载γ-MnO2纳米复合材料活化过一硫酸盐降解对氯苯酚的性能及机理[J]. 环境工程学报, 2022, 16(6): 1833-1844. doi: 10.12030/j.cjee.202201078 [12] SHEN S, ZHOU X, ZHAO Q, et al. Understanding the nonradical activation of peroxymonosulfate by different crystallographic MnO2: The pivotal role of MnIII content on the surface[J]. Journal of Hazardous Materials, 2022, 439: 129613. doi: 10.1016/j.jhazmat.2022.129613 [13] HUANG J, DAI Y, SINGEWALD K, et al. Effects of MnO2 of different structures on activation of peroxymonosulfate for bisphenol a degradation under acidic conditions[J]. Chemical Engineering Journal, 2019, 370: 906-915. doi: 10.1016/j.cej.2019.03.238 [14] WANG Y, ZHONG W, ZHANG S, et al. Pearl necklace-like comn-based nanostructures derived from metal-organic frames for enhanced electromagnetic wave absorption[J]. Carbon, 2022, 188: 254-264. doi: 10.1016/j.carbon.2021.12.030 [15] WU S, WANG C Z, JIN Y Q, et al. Green synthesis of reusable super-paramagnetic diatomite for aqueous nickel (II) removal[J]. Journal of Colloid and Interface Science, 2021, 582: 1179-1190. doi: 10.1016/j.jcis.2020.08.119 [16] QI X, XIE F. Promotion effects of potassium permanganate on removal of Pb(II), Ni(II) and Cd(II) from hydrous manganese dioxide[J]. Chemical Engineering Journal, 2018, 351: 22-30. doi: 10.1016/j.cej.2018.06.042 [17] HU P, SU H, CHEN Z, et al. Selective degradation of organic pollutants using an efficient metal-free catalyst derived from carbonized polypyrrole via peroxymonosulfate activation[J]. Environmental Science & Technology, 2017, 51(19): 11288-11296. [18] WANG Y, INDRAWIRAWAN S, DUAN X, et al. New insights into heterogeneous generation and evolution processes of sulfate radicals for phenol degradation over one-dimensional α-MnO2 nanostructures[J]. Chemical Engineering Journal, 2015, 266: 12-20. doi: 10.1016/j.cej.2014.12.066 [19] RUIZ-CAMACHO B, BALTAZAR VERA J C, MEDINA-RAMíREZ A, et al. Eis analysis of oxygen reduction reaction of pt supported on different substrates[J]. International Journal of Hydrogen Energy, 2017, 42(51): 30364-30373. doi: 10.1016/j.ijhydene.2017.08.087 [20] SINGH R K, DEVIVARAPRASAD R, KAR T, et al. Electrochemical impedance spectroscopy of oxygen reduction reaction (ORR) in a rotating disk electrode configuration: Effect of ionomer content and carbon-support[J]. Journal of the Electrochemical Society, 2015, 162(6): F489-F498. doi: 10.1149/2.0141506jes [21] SAPUTRA E, MUHAMMAD S, SUN H, et al. Different crystallographic one-dimensional MnO2 nanomaterials and their superior performance in catalytic phenol degradation[J]. Environmental Science & Technology, 2013, 47(11): 5882-5887. [22] MA J, ZHANG S, DUAN X, et al. Catalytic oxidation of sulfachloropyridazine by MnO2: Effects of crystalline phase and peroxide oxidants[J]. Chemosphere, 2021, 267: 129287. doi: 10.1016/j.chemosphere.2020.129287 [23] 何锦垚, 魏健, 张嘉雯, 等. 臭氧催化氧化-BAF组合工艺深度处理抗生素制药废水[J]. 环境工程学报, 2019, 13(10): 2385-2392. doi: 10.12030/j.cjee.201902043 [24] BENITEZ F J, ACERO J L, REAL F J, et al. Ozonation of benzotriazole and methylindole: Kinetic modeling, identification of intermediates and reaction mechanisms[J]. Journal of Hazardous Materials, 2015, 282: 224-232. doi: 10.1016/j.jhazmat.2014.05.085 [25] 王业耀 王占生. 影响臭氧化过程的水质指标[J]. 中国给水排水, 1996(6): 29-31. doi: 10.3321/j.issn:1000-4602.1996.06.010 [26] CHEN X, DENG F, LIU X, et al. Hydrothermal synthesis of MnO2/Fe(0) composites from li-ion battery cathodes for destructing sulfadiazine by photo-fenton process[J]. Science of the Total Environment, 2021, 774: 145776. doi: 10.1016/j.scitotenv.2021.145776 [27] WANG H, GUO W, LIU B, et al. Edge-nitrogenated biochar for efficient peroxydisulfate activation: An electron transfer mechanism[J]. Water Research, 2019, 160: 405-414. doi: 10.1016/j.watres.2019.05.059 [28] LUO M, ZHOU H, ZHOU P, et al. Insights into the role of in-situ and ex-situ hydrogen peroxide for enhanced ferrate(VI) towards oxidation of organic contaminants[J]. Water Research, 2021, 203: 117584. [29] ZHAO Z, ZHOU W, LIN D, et al. Construction of dual active sites on diatomic metal (FeCo−N/C-x) catalysts for enhanced fenton-like catalysis[J]. Applied Catalysis B: Environmental, 2022, 309: 121256. doi: 10.1016/j.apcatb.2022.121256 [30] WANG Y, DUAN X, XIE Y, et al. Nanocarbon-based catalytic ozonation for aqueous oxidation: Engineering defects for active sites and tunable reaction pathways[J]. ACS Catalysis, 2020, 10(22): 13383-13414. doi: 10.1021/acscatal.0c04232 [31] ZHOU C, ZHOU P, SUN M, et al. Nitrogen-doped carbon nanotubes enhanced fenton chemistry: Role of near-free iron(Ⅲ) for sustainable iron(Ⅲ)/iron(II) cycles[J]. Water Research, 2022, 210: 117984. doi: 10.1016/j.watres.2021.117984 [32] XU L, FU B, SUN Y, et al. Degradation of organic pollutants by Fe/N co-doped biochar via peroxymonosulfate activation: Synthesis, performance, mechanism and its potential for practical application[J]. Chemical Engineering Journal, 2020, 400: 125870. doi: 10.1016/j.cej.2020.125870 [33] SONG Z, ZHANG Y, LIU C, et al. Insight into oh and O2− formation in heterogeneous catalytic ozonation by delocalized electrons and surface oxygen-containing functional groups in layered-structure nanocarbons[J]. Chemical Engineering Journal, 2019, 357: 655-666. doi: 10.1016/j.cej.2018.09.182 [34] ZHANG Y, JI H, LIU W, et al. Synchronous degradation of aqueous benzotriazole and bromate reduction in catalytic ozonation: Effect of matrix factor, degradation mechanism and application strategy in water treatment[J]. Science of the Total Environment, 2020, 727: 138696. doi: 10.1016/j.scitotenv.2020.138696 [35] OUYANG C, WEI K, HUANG X, et al. Bifunctional fe for induced graphitization and catalytic ozonation based on a Fe/N-Doped carbon–Al2O3 framework: Theoretical calculations guided catalyst design and optimization[J]. Environmental Science & Technology, 2021, 55(16): 11236-11244. [36] WANG D, HE Y, CHEN Y, et al. Electron transfer enhancing the Mn(II)/Mn(Ⅲ) cycle in MnO/CN towards catalytic ozonation of atrazine via a synergistic effect between MnO and CN[J]. Water Research, 2023, 230: 119574. doi: 10.1016/j.watres.2023.119574 -
 点击查看大图
点击查看大图

计量
- 文章访问数: 1186
- HTML全文浏览数: 1186
- PDF下载数: 41
- 施引文献: 0
