

-
高级氧化技术(AOPs,advanced oxidation processes)是一种以产生具有强氧化能力的羟基自由基(·OH)为特点,将难降解有机物氧化成小分子物质或完全矿化的污染物处理技术[1-2]。近年来,高级氧化水处理技术得到了较快发展,广泛应用于工业废水处理、土壤和地下水修复、污水深度处理等。通过激活氧化剂(如臭氧、过氧化氢、过硫酸盐等)生成活性自由基,可将有机物逐步降解为小分子物质,如二氧化碳、水和无机盐等,甚至完全矿化,达到高效去除有毒有害污染物的目的[3-5]。
芬顿法是高级氧化工艺(AOPs)的一种,其通过亚铁离子和过氧化氢反应,生成羟基自由基。羟基自由基具有极强的氧化性,能有效降解有机污染物[6-7]。传统的芬顿反应工艺优势明显,但仍存在诸多缺陷,如pH响应范围窄(pH 2.8—3.5最佳)、铁泥产量大、H2O2利用率低等[8]。芬顿氧化技术对有机物的降解效率与反应体系的pH密切相关。在氧化还原体系中,H+和OH−离子影响铁的水解形态,进而影响芬顿反应。当pH>3.5时,溶解性Fe2+形成氧化物、氢氧化物等物种,致使催化活性降低;而当pH过低时,H2O2质子化形成H3O2+,无法与铁物种反应产生·OH,这些因素限制了传统芬顿反应的适用范围。在芬顿反应过程中,不可避免地会产生体系pH的波动,因而铁物种沉淀导致产生大量铁泥。除这些缺点外,传统芬顿反应H2O2的利用率低也是限制芬顿技术应用的一大难题。在反应中,Fe3+与H2O2反应产生催化活性很低的HO2·,并会进一步与·OH反应使其淬灭,影响了整个体系的降解能力。
为了克服均相芬顿反应的缺陷,研究人员将目光聚焦于催化剂及激活氧化剂两方面。一方面,研究人员使用其他过渡金属作为均相芬顿反应的催化剂,如:Cu2+,其溶解的pH值范围在3—7[9-10]。另外,研究人员采用固体催化剂活化H2O2,使得整个非均相芬顿体系不产生铁泥,且可适用于较宽的pH范围,克服了传统均相芬顿的缺点[7, 11]。另一方面,过氧二硫酸盐(PDS)和过氧一硫酸盐(PMS)在废水处理中也受到越来越多研究人员的关注。过硫酸盐是强氧化剂,能够直接降解有机污染物。但是过硫酸盐与H2O2类似,其与大多数有机污染物的直接反应很慢,因此,在应用中通常将过硫酸盐进行活化,产生氧化性更强的SO4−·或·OH。SO4·−具有高的氧化还原电势,其自由基寿命比·OH长,可在宽pH(2—12)范围内应用[12]。在过硫酸盐的活化过程中,部分活化剂的表面氧化还原电势驱动单电子转移的链反应,以裂解过氧化物键形成SO4·−和·OH,并通过这些瞬态自由基氧化降解有机污染物[13]。
与活化过氧化氢的芬顿系统相比,过硫酸盐类芬顿系统涉及了更复杂的氧化途径,除了存在自由基氧化途径外,还存在着多种非自由基氧化途径(如单线态氧1O2),其氧化途径取决于水质和活化剂的类型。过硫酸盐被碳基材料[14-15],N掺杂的石墨烯[16-17],硼掺杂有序介孔碳[18],生物炭[19]等活化时,某些氧化路径不仅涉及自由基,还有非自由基1O2,在中性条件下即可选择性氧化有机物。除此之外,还有文献报道碳纳米管活化过硫酸盐的非自由基途径并非单线态氧,而是通过碳形成的络合物介导电子从有机物转移至过硫酸盐,从而使有机物降解[15, 20-23]。
本文综述了类芬顿催化剂的设计、催化活化原理与机制等方面的研究进展,介绍了基于过渡金属及其氧化物的催化剂,及其在水处理中降解有机污染物的机理,并提出了存在的问题及研究展望。
-
在类芬顿系统中,对过氧化氢和过硫酸盐的催化,常使用低价过渡金属及其改性材料作为催化剂。与均相芬顿相比,非均相催化剂以不产生铁泥,固液易分离,且可以在较宽的pH范围应用等优势成为较有前景的替代方案[7, 11]。本文将非均相芬顿催化剂分为铁基、锰基、钴基、铜基催化剂和负载型催化剂,并论述了各类催化剂的设计原理、性能、催化机理以及作用机制。
-
过氧化氢和过硫酸盐的活化主要通过低价态过渡金属,金属氧化物和某些复合物、聚合物来实现。用作铁基非均相芬顿催化的主要是铁氧化物或金属复合氧化物,其中铁元素固定在结构中,避免了铁离子的大量溶出,使催化剂在较宽pH范围内稳定存在。例如,磁铁矿具有良好的氧化还原性、结构稳定性以及磁性,是常见的非均相芬顿催化剂[24]。天然磁铁矿通过表面物种(Fe2+和Fe3+)可逆的氧化还原来活化H2O2产生·OH降解污染物(反应式1和2)。
天然磁铁矿存在表面羟基(≡Fe—OH)作为活性催化位点,≡Fe-OH可以吸附H2O2产生∙OH[25]。含有镁铁尖晶石固溶相的天然磁铁矿为面心立方晶胞,可同时容纳Fe2+和Fe3+,其中Fe2+占据八面体位点,Fe3+占据四面体和八面体位点。这种特征使得Fe2+被可逆地氧化和还原,同时保留原始晶体结构,且加速内部电子转移,使催化剂具有优异的异质催化能力[24,26-27]。此外,有研究认为在天然磁铁矿中,杂金属原子的同构取代能提高磁铁矿的催化效率。早期研究报道,Co和Mn的存在可有效改善磁铁矿的还原能力,使催化活性显著提高,而Ni则抑制H2O2的活化[28]。有研究报道称,磁铁矿中Fe2+被氧化会导致还原能力丧失,此时若水中有Fe2+(aq),则Fe2+会吸附到磁铁矿上并将电子转移到氧化的磁铁矿上,使八面体中的Fe3+还原为Fe2+,这种磁铁矿与Fe2+(aq)耦合系统,可使磁铁矿形成持续的催化能力[29]。研究人员合成了与天然磁铁矿具有相似取代水平(x≈0.5)的磁铁矿(Fe3-xMxO4,M = Co2+、Mn2+、Zn2+、Mg2+、Cr3+和Al3+),发现Co、Mn、Zn和Mg可以显著提升磁铁矿与Fe2+(aq)耦合系统的还原能力(图1a)[30-31]。这是由于Co,Mn,Zn和Mg等会增加活性位点密度,且八面体Co和Mn具有氧化还原对(即Co2+ / Co3+和Mn2+ / Mn3+),四面体Zn2+诱导八面体Fe2+氧化为Fe3+,可加速电子转移,从而导致电导率的增加,提高其催化活化性能。过渡金属如Cu的掺杂可以形成Cu和Fe3O4间的肖特基界面,使催化剂具备良好的电子供体特征,且Fe3+ / Fe2+循环和结构缺陷加速了电荷转移过程,促进∙OH和O2·−自由基的形成,大大提升了有机污染物的降解效率(如图1b)[32]。
在Fe3O4对H2O2的催化活化过程中,涉及Fe3+ / Fe2+的氧化还原异相芬顿过程,也存在溶液中的均相芬顿过程,具体取决于pH值和催化剂的性质[33]。在较低pH值下,均相、异相反应同时发生;随着pH值增大,异相芬顿过程占据主导地位[17]。Wang等[34]报道通过在Fe3O4上涂覆pH敏感的共聚物水凝胶合成的Fe3O4磁性复合纳米球,使磁铁矿颗粒稳定分散,且催化剂在酸或强碱溶液中不易损坏,因此可在宽pH范围下进行有效催化。因此,如图1c所示,由于延长的聚合物链存在空间位阻效应,防止了纳米粒子的聚集,Fe3O4 MCN可以在pH 3和pH 9之间稳定地分散。
-
锰(Mn)元素有从0至+7的多种价态,天然的锰物种主要以氧化物形式(Mn3O4、Mn2O3和MnO2)存在,而在水环境中多数以Mn2+或Mn4+的形式存在[35]。锰氧化物在催化活化过氧化氢和过硫酸盐过程中起重要作用,可将其活化生成不同类型的活性物种,包括SO4·−、·OH、HO2·、O2·−和1O2等[36]。锰基催化剂活化过硫酸盐的活性顺序通常为MnO2≥Mn2O3>Mn3O4,活化能力与催化剂的氧迁移率、形态、晶体类型和比表面积等相关[37-38]。Liu等[39]提出,在活化过硫酸盐反应中,≡Mn(Ⅳ)的单电子还原导致过硫酸根自由基(S2O8·−)形成,从而引发自由基链反应。随后≡Mn(Ⅲ)被过硫酸盐快速氧化,产生SO4·−和SO42-,同时其自身形成≡Mn(Ⅳ)。此过程中,过硫酸盐既可被氧化又可被还原,这与芬顿过程中H2O2既可作氧化剂又可作还原剂的机制是一致的。活化生成的SO4·−还可与H2O或OH−继续反应生成OH·。锰的多种价态有利于这种氧化还原过程的发生。例如,氧化锰八面体分子筛(OMS-2)是二氧化锰的一种形式,其包含混合价态锰(Mn2+、Mn3+和Mn4+)[40]。通过高价态锰氧化物(Mn4+)还原和低价态锰氧化物(Mn3+)氧化可以活化PMS生成SO4−·和SO5−·,其中SO4−·与H2O的反应生成∙OH[41]。从Mn3+到Mn4+的转变在热力学上有利于激活PMS以生成SO4∙− 和∙OH(反应式3和4)。类似地,也可以将Mn4+转化为Mn3+以进一步产生∙OH(反应式5和6),部分∙OH被PMS诱导产生SO4∙−(反应式7)[38]。
研究发现,MnO2比Mn2O3和Mn3O4具有更高的反应活性。具有较低价态的Mn2O3和Mn3O4催化剂的活性却较低,表明PDS活化中的锰不只是简单地作为电子供体。如在β-MnO2活化PDS过程中,除了SO4∙−和∙OH外,还生成了单线态氧(1O2),并且以其作为主要活性氧物种(如图2a),1O2可能是由MnO2表面的亚稳态锰中间体产生的超氧自由基直接氧化或重组产生[42]。α-MnO2、β-MnO2的1O2有相同催化路径,1O2对有机污染物的氧化是在体相溶液而不是在催化剂表面进行的[43]。MnO2由于晶型差异也显示出不同的反应活性,基本表现为α-MnO2>γ-MnO2>β-MnO2。这是由于α-MnO2、γ-MnO2的MnO6八面体共享边缘暴露多,并且α-MnO2具有Mn4+→Mn3+→Mn2+的氧化转变过程,而γ-MnO2只能由Mn4+转变为Mn3+共同导致的[37]。由此可以看出,锰氧化物活化PS并非只涉及自由基途径,而是以非自由基途径为主导,且其晶体结构及氧化还原电势对其催化活化性能也有较大的影响。
通过掺杂金属可以改变锰氧化物的表面和晶体结构,从而提高反应活性和催化剂稳定性。经过渡金属掺杂的锰氧化物,金属物种之间的氧化还原循环发生改变,形成催化活性位点,增强了与过氧化物分子间的相互作用并增加了吸附位点。例如,CoMn2O4在活化PMS过程中表现出比Mn2O3和Co3O4及其物理混合物更强的催化活性。由于钴的掺杂,CoMn2O4中Co和Mn物种之间形成协同效应,形成了≡CoⅢ/CoⅡ、≡MnⅢ/MnⅡ、≡MnⅢ/CoⅡ和≡MnⅣ/MnⅢ等多种氧化还原电对,使PMS活化率增高。≡Co和≡Mn活化PMS生成·OH,而≡Co-−OH和≡Mn-−OH活化PMS生成表面结合态的SO4−·[23] (如图2b)。铁掺杂的锰氧化物中界面铁锰双活性中心通过电荷重排加速电子传输,通过PMS的亲电子氧化,生成1O2或直接氧化有机污染物(如图2c)。由于在可逆氧化还原循环中结构能保持稳定,铁锰双活性中心结构可保持持久的催化协同效果[44]。此外,其他金属元素如Ce掺杂的Mn2O3因能生成更多的还原性金属物种,产生更多的氧空位,促进界面电子转移,利于PMS活化生成∙OH和SO4·−,提升有机污染物的降解效率(如图2d)[45]。
-
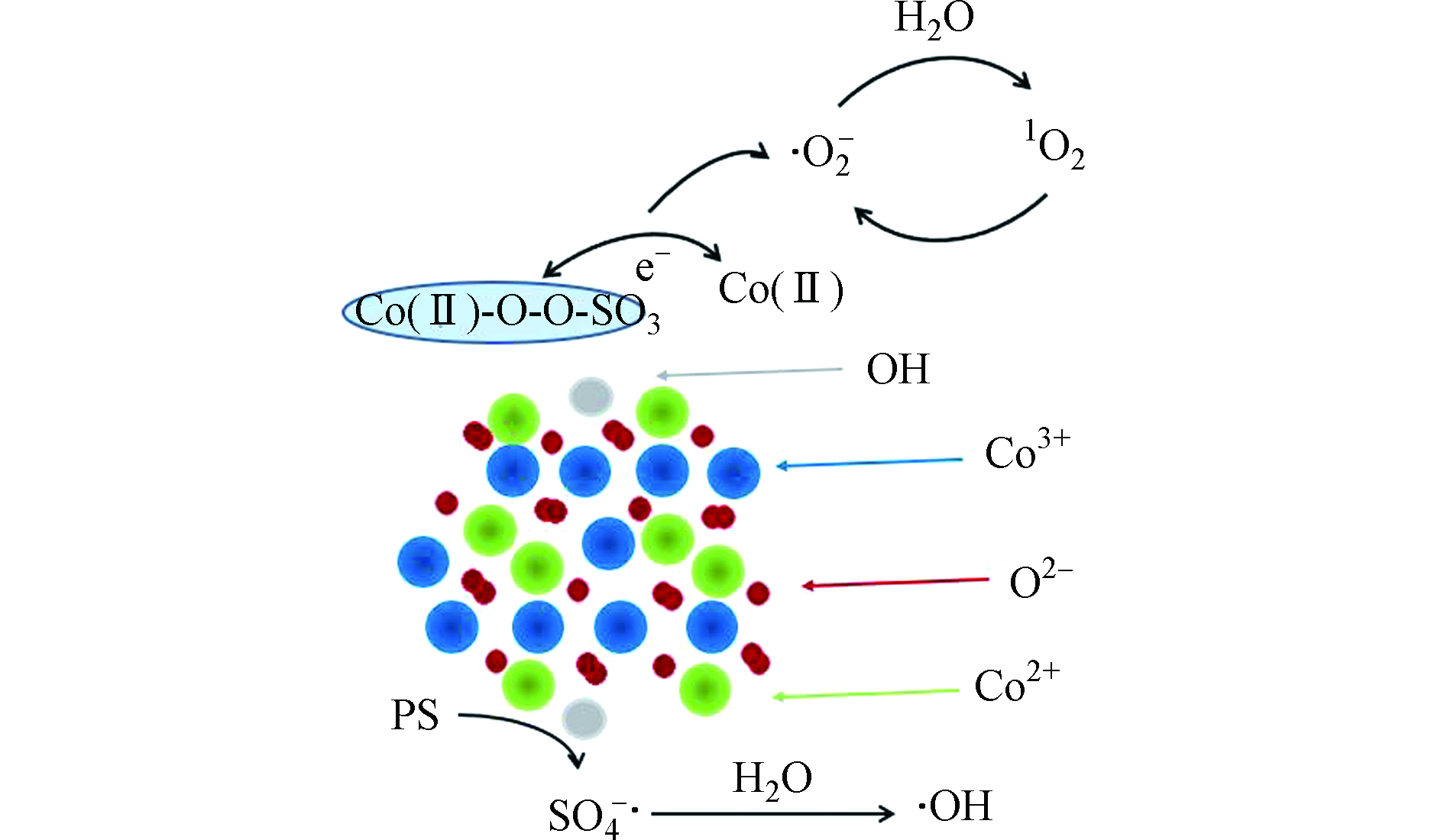
在过渡金属元素中,Co是活化过硫酸盐的高效催化剂之一[46]。与所有其他过渡金属相比,Co2+对过硫酸盐的催化活化效率最高,在催化反应过程中,可持续的进行Co2+的自身循环完成过硫酸盐的活化[47]。有研究曾将多种金属离子与普鲁士蓝类配合物(PBA)共同制备形成MII3[MIII(CN)6]2 (MII = Co, Cu, Fe, Mn, Ni; MIII =Co, Fe),发现Co的PBA催化活性远高于其他金属[48]。针对Co的催化活化研究,大多数都集中在使用Co2+/Co3+氧化还原对[E0(Co3+/Co2+)= +1.92 V]来活化过硫酸盐,生成SO4∙−作为主要的氧化剂(反应式8和9)。
钴基材料中,钴氧化物或掺杂其他过渡金属的钴氧化物是常见的催化剂[49]。钴氧化物表面形态和结构对过硫酸盐的催化活性影响较大,其催化活性顺序为纳米立方体(NC)>花状纳米板(FN)>纳米片(NS)>薄片堆叠的微球(FM)[50],由此发现具有(111)暴露晶面和更多表面羟基的纳米立方体样品在过硫酸盐活化过程中对有机污染物的去除表现出最高的催化活性。同其他过渡金属类似,钴基材料的活性位点主要是≡Co和≡Co-OH。例如,报道中提出,污染物降解的过程既包括自由基又包括非自由基路径,且非自由基(形成1O2)路径起了关键作用(如图3)[50]。这种活化方式与前文提到的锰氧化物活化过硫酸盐机理保持一致,均是通过与H2O离解吸附耦合,Co3O4表面生成CoOH+(反应式10),而CoOH+可以通过H键与S2O82−结合,Co2+被氧化成Co3+(CoO+),形成亚稳态钴中间体,O—H和O—O接受电子后同时断裂,生成高反应活性的SO4·−(方程式11),SO4·−可进一步与H2O反应生成·OH(反应式12)。CoO+可催化S2O82−生成O2·−,而H2O与O2·−可以反应生成1O2(反应式13和14)。
掺杂过渡金属的钴氧化物可通过双金属或多金属协同实现对过氧化氢和过硫酸盐的活化。Tatarchuk等[51]研究人员合成了具有尖晶石结构的CoCr2-xFexO4(x=0.0—2.0),其中铬离子主要位于八面体位置,而钴和三价铁离子分布在八面体和四面体位置之间。在整个体系中,Fe是催化活化H2O2的活性中心,而Co2+ / Co3+氧化还原对可电子转移从而提高整个体系的催化活性。非晶态结构的Fe36Co36Si4.8B19.2Nb4金属玻璃(FeCoSiBNb MG),也可以使得Fe和Co元素作为电子供体通过四电子转移机制活化过硫酸盐[52]。在多金属层状双氧化物(CoCuAl—LDOs)催化过硫酸盐过程中,钴铜协同起到了促进Co2+ / Co3+氧化还原循环的作用,加快了SO4∙−的生成,从而提升了催化效率[53]。尽管钴是优质的过氧化物催化剂,但存在严重的缺陷,即钴的毒性较强,异质芬顿催化剂发生钴离子浸出会对环境造成很大危害,这影响了钴基材料作为催化剂的应用。
-
铜具有多种化合价(Cu0,Cu+和Cu2+),且这3种铜物种都可以活化H2O2分解以产生活性氧物种[54]。Cu0会失去一个电子而生成Cu+(反应式15),Cu2+可被H2O2还原为Cu+(反应式16),然后与H2O2进一步反应形成·OH(反应式18)。
有研究认为Cu2+被还原为Cu+生成的是O2·−(反应式17),且认为O2·−是有机污染物降解的主要活性物质[55]。由于Cu2+向Cu+转化是·OH生成的限速步骤,因此Cu+是非均相芬顿反应的主要催化活性位。
为了充分利用铜基芬顿系统的潜力,研究人员用其他金属与铜形成双金属或多金属复合物,以实现更高的催化活性、适用性和化学稳定性。如前所述,Cu+在非均相芬顿反应中占主导作用,因此通过掺杂金属(如稀土元素铈)来调节金属催化剂上Cu+的相对含量,同时改变电荷分布,使得催化剂表面形成富电中心加速·OH的生成,可提高整体反应的催化活性[56]。有研究将钒引入铜基材料中,铜、钒活性位周围形成富电中心和表面氧空位,也可加快H2O2的离解和活化(如图4a)[57]。Chen等[58]合成了铜基多金属层状双氢氧化物(CuNiFeLa—LDHs)(如图4b)来降解水中有机污染物。该催化剂以Cu为主要催化活性位点,其中Ni2+可以将电子转移至Cu2+(反应式19),通过金属—氧—金属(M—O—M)结构生成更多的Cu+。La的掺杂可制造出更多的孔结构,使H2O2和有机污染物更容易进入催化剂中。La2O3可同时提供Lewis碱性位点和Lewis酸性位点,为H2O2的催化活化提供了更多的反应活性位。在整个系统中,除了Cu参与H2O2的活化外,还涉及以下反应机制:
Wang等[59]合成了嵌入Cu-Co双金属纳米颗粒的碳纳米立方体Cu4Co6 / CNC。在活化H2O2过程中,Cu4Co6 / CNC中的Co0和Cu0被氧化为Co2+和Cu+。Co2+和Cu+活化H2O2产生·OH,且Cu+可以将生成的Co3+还原为Co2+,实现了Co2+的再生。除了铜对于催化活性的促进外,碳骨架也可以增强Co / Cu纳米粒子的分散性和稳定性,促进催化反应的电子转移,并提供更多的催化活性中心。
在上述研究中,铜可以在很宽的pH范围下发生芬顿反应。在酸性条件下,铜的溶出量较大,而中性条件下更稳定,这有利于铜基催化剂的实际应用。但也有研究表明,在酸性和近中性条件下,Cu+会被氧气氧化成Cu2+,特别是在以Cu+为活性位的催化体系中,H2O2的有效利用率会降低[60]。胡春等[61]合成出一种新型高效双反应中心催化剂OH—CCN / CuCo—Al2O3。该催化剂形成了富电子铜中心和缺电子氮中心,H2O2接受富电子铜中心的电子形成·OH,同时H2O的电子转移到缺电子氮中心,也能转化为·OH,从而极大地提高了H2O2的有效利用率。且缺电子中心获得的电子会通过C—O—Cu键桥传递到富电子铜中心,从而维持整个系统电子得失平衡(图4c)。
对于铜活化过硫酸盐过程,已提出了两种主要机制:自由基路径与非自由基路径。Lei等[62]研究磁性CuO—Fe3O4催化剂时提出,过硫酸盐活化时CuO-Fe3O4表面形成络合物(反应式21),该络合物可随即分解出SO4·−(反应式22),而生成的Cu3+,在没有强螯合阴离子的存在下不稳定,会生成·OH(反应式23)。
对于非自由基路径,Jawad等[63]在研究CuOMgO / Fe3O4时,认为在CuOMgO / Fe3O4表面形成表面羟基,该物质与过硫酸盐反应,生成亚稳态的铜中间体(反应式25)。该中间体得到另一个过硫酸盐的电子后失稳,生成O2·−(反应式26)。随后,亚稳态中间体被O2·−氧化生成1O2(反应式27),1O2在降解污染物过程中起主导作(如图4d)。
-
碳材料(如活性炭、碳纤维、碳气凝胶、碳纳米管和石墨烯等)常被用作非均相芬顿催化剂载体。活性炭具有较大的比表面积、高孔隙率和各种官能团,有利于其吸附有机污染物[64]。同时,多孔结构使得催化剂在材料表面和内部高度分散,改善了反应活性位点数量和性质,有助于实现活性炭吸附和芬顿生成的·OH氧化协同作用[65]。Wang等[66]合成了一系列具有不同铁/碳比的铁氧体气凝胶(FCA),FCA表现出良好的电导率,高催化效率和出色的稳定性。多孔FCA的活性成分是Fe与α-Fe2O3的混合相,由于电子中从Fe0到Fe3+的电子转移形成了Fesurf2+,使得催化剂保持高含量Fe2+,从而保持高催化活性。
Yao等[67]研究了石墨烯纳米片 / FexOy / 氮掺杂碳气凝胶复合催化剂,提出了Fe—O—C结构可加速Fe3+ / Fe2+的氧化还原循环,从而催化剂具有良好的催化活性和可重复使用性。Chen等[68]在碳纳米管表面负载了Al0,Fe0和Cu0颗粒(图5a),实现了中性pH条件下芬顿反应的高活性。其中Al0-CNTs起活化O2的作用,而CNTs—Fe—Cu不仅能活化O2,还可以将H2O2催化生成·OH和O2·−。Liu等[69]研究了碳纳米管负载Co3O4催化剂,认为Co3O4 @ CNT具有良好的导电性,且Co3O4中的Co具有独特的价态组成结构,使其能够在Co2+和Co3+之间产生单电子转移,促进·OH和SO4·−的形成。而在Fe2O3 @ FCNT—H / H2O2系统中,研究者认为H2O2是生成1O2的唯一来源,H2O2吸收两个电子生成1O2(如图5b),最终达到氧化有机污染物的目的[70]。
分子筛具有精确而均匀的孔道结构,常作为优异的催化剂或载体被广泛应用[71-72]。Mazilu等[73]合成了含铁的SBA-15催化剂,在弱碱性的pH值下,铁元素以高度分散的形式掺入了中孔SBA-15结构中,且铝与铁的共添加显著促进了铁离子在高度分散状态下的稳定化,提高了系统的催化活性。Yin等[74]将单原子Fe负载在SBA-15上以得到暴露最大的Fe活性位,发现单原子铁催化剂比聚集的铁催化剂展现出更高的活性。Sun等[75]合成了双金属FeCo掺杂的MCM-41,发现分散后的钴和铁之间的协同氧化还原循环也是高催化活性的原因(图5c)。综上可知,负载型催化剂的载体电子转移能力强,催化剂高度分散在载体表面,可以加速金属的氧化还原循环,显著提高芬顿反应性。
-
如前所述,在类芬顿体系中,过渡金属及其氧化物通过催化活化过氧化氢或过硫酸盐,形成强氧化性自由基以降解有机污染物。过氧化氢和过硫酸盐的催化活化原理与路径受催化剂、水质条件等因素影响。其中过氧化氢的催化活化主要通过增加表面活性位点、金属掺杂及多金属协同等作用强化催化活化效率(表1),以增加过氧化氢的利用率、扩大pH适用范围和降低副产物(如铁泥)的生成量。
过硫酸盐的活化过程相对复杂,反应路径和活性物种类型较过氧化氢催化多(表2)。金属物种引发过硫酸盐单电子还原产生SO4−·,与·OH相比氧化性能相当,且在特定条件下与水中氢氧根离子反应生成·OH。SO4−·的寿命更长,与有机污染物持续接触以延长氧化时间,可处理部分羟自由基自身无法氧化的有机物。在实际应用中,须根据目标污染物和应用场景选择合适的催化材料和反应条件。而活化过硫酸盐对于污染物的降解,除了涉及自由基途径,还存在非自由基途径(1O2),且其为氧化有机污染物的重要途径。可以通过以过硫酸盐作供氧体,与催化剂形成高氧化性金属过氧物种(亚稳态中间体),中间体从其他过硫酸根处得一个电子后生成O2·−,最终反应产生1O2,用以有机污染物的氧化[32, 37, 63]。
-
非均相类芬顿催化体系通过过渡金属及其氧化物将过氧化氢或过硫酸盐等催化活化形成强氧化性的自由基以及非自由基,以达到降解及矿化有机污染物的目的。针对非均相芬顿反应存在的问题和缺点,许多研究提出了多种提高非均相芬顿反应体系效率的方式,如控制催化剂的形貌和暴露面、金属掺杂、增加表面活性位点、多金属协同、构建富电子中心等。通过改变金属的形态及价态,改变催化剂物理化学结构以增加活性中心;引入其他金属实现多金属协同作用,或加入某些物质,使其与催化剂形成利于污染物降解的中间体结构,促进非均相类芬顿催化反应。目前,非均相类芬顿体系的机理研究虽然已取得了一定进展,但要实现大规模应用尚存在许多问题需要解决。在今后发展过程中,需要进一步探究金属基催化剂催化活化性能提高的新途径,并通过金属及催化剂催化机理指导发展非金属基催化剂在非均相类芬顿体系中的应用。
类芬顿反应的催化剂、原理与机制研究进展
Research progress on catalysts, principles and mechanisms of Fenton-like reactions
-
摘要: 芬顿氧化法是高级氧化技术的一种,通过产生高活性的自由基降解水中有机污染物,在水处理领域受到广泛关注。近年来,针对芬顿反应的研究主要集中在开发高活性、高稳定性的非均相类芬顿催化剂。本文综述了近年来非均相芬顿催化领域的研究进展,分析了铁基、锰基、铜基、钴基等过渡金属及其氧化物,以及负载型催化剂的构造特点、催化机理和作用机制,探讨了过氧化氢和过硫酸盐的催化活化路径与方式,总结了非均相类芬顿催化体系实现高效降解有机污染物的机理与途径,以期为高效类芬顿催化剂的设计和应用提供科学依据.Abstract: Fenton processes is an advanced oxidation technology, which degrades organic pollutants by generating highly active free radicals, and has gained much attention in the field of wastewater treatment. In recent years, research on Fenton reaction has focused more on the development of heterogeneous Fenton-like catalysts with high activity and high stability. This article reviews the research progress in the field of heterogeneous Fenton catalysis in recent years, and analyzes the structure characteristics and catalytic mechanism of iron-based, manganese-based, copper-based, cobalt-based and other transition metals and their oxides, as well as supported catalysts. Extensive discussion on the catalytic pathways of hydrogen peroxide and persulfate, and the mechanism and pathways of the heterogeneous Fenton-like catalytic system to achieve high-efficiency degradation of organic pollutants were provided along with literature of some relevant studies, in order to provide a scientific basis for the design and application of efficient Fenton-like catalysts.
-
Key words:
- advanced oxidation /
- heterogeneous Fenton /
- Fenton-like reaction /
- hydrogen peroxide /
- persulfate
-

-
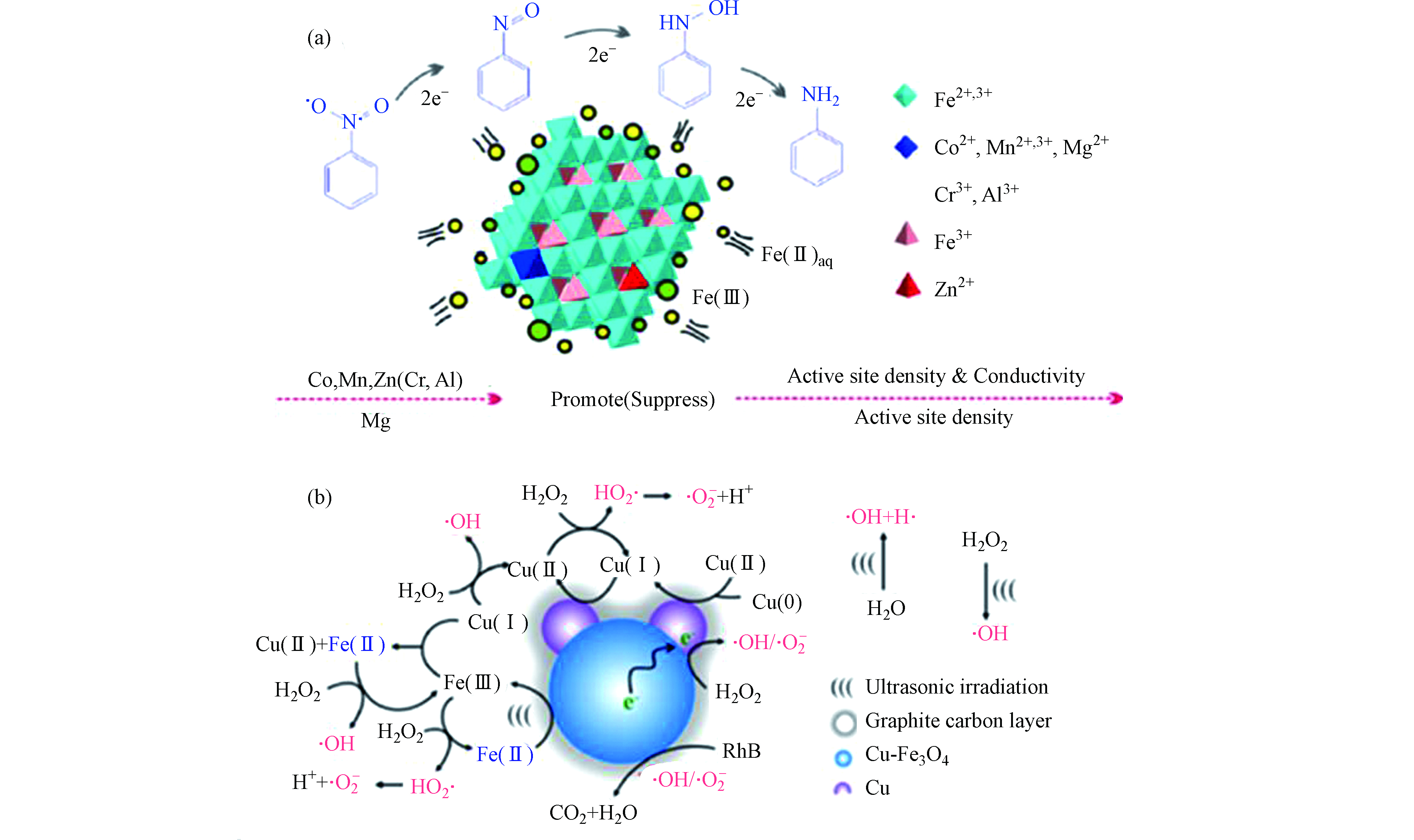
图 1 (a) 磁铁矿Fe3-xMxO4(M = Co2+,Mn2+,Zn2+,Mg2+,Cr3+和Al3+)耦合Fe2+芬顿氧化降解污染物[31],(b) Cu—Fe3O4/Cu/C活化过氧化氢示意图[32]
Figure 1. (a) The degradation of organic pollutants by Fenton oxidation with magnetite Fe3-xMxO4(M = Co2+, Mn2+, Zn2+, Mg2+, Cr3+ and Al3+) coupled Fe2+ [31], (b) The diagram of Cu-Fe3O4/Cu/C in hydrogen peroxide activation [32]
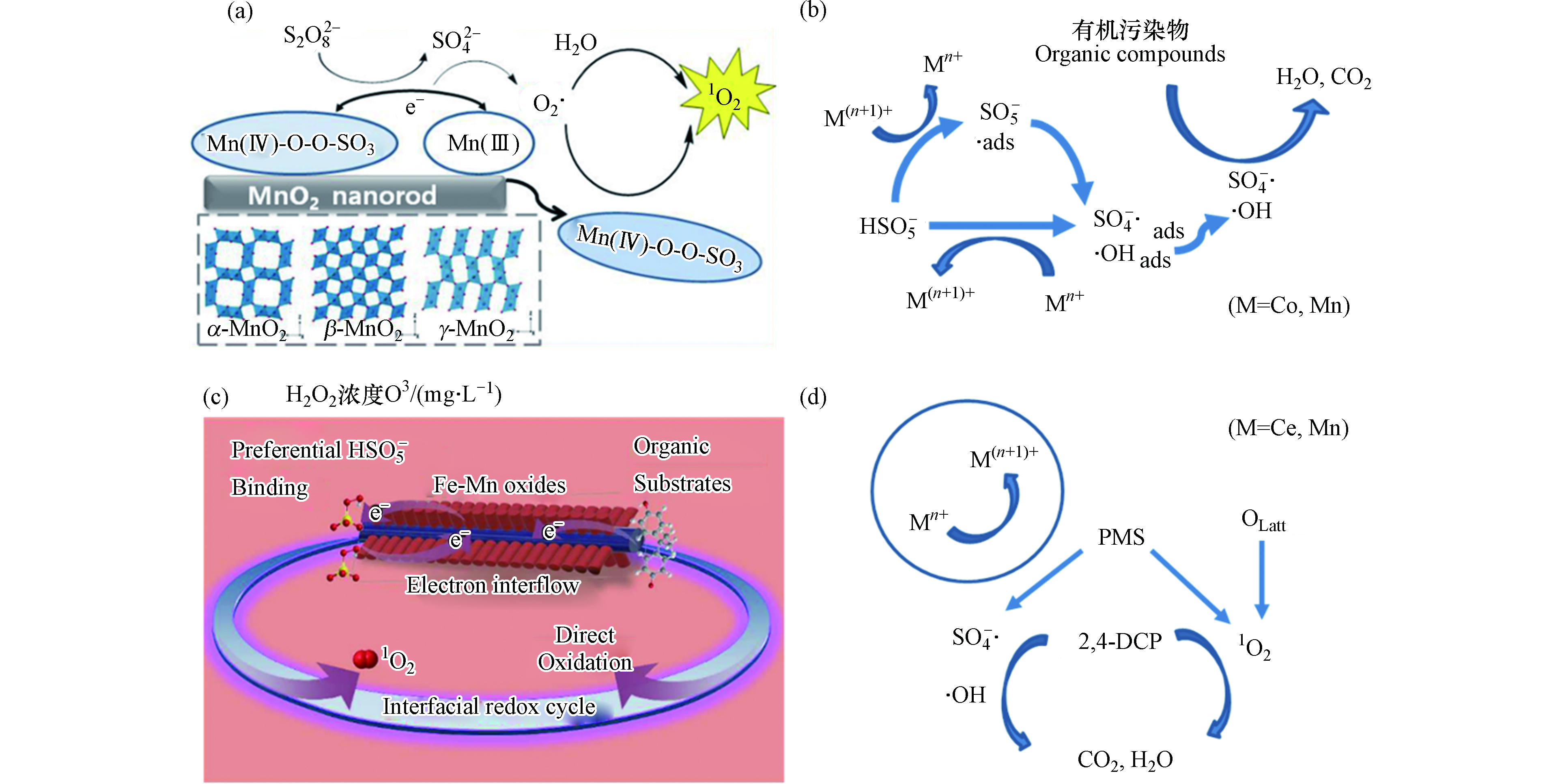
图 2 (a) MnO2晶体活化PDS生成1O2的反应机理[42],(b) Co—Mn协同活化PMS机理图[23],(c) Fe掺杂的锰氧化物活化PMS机理图[44],(d) Ce掺杂锰氧化物活化PDS机理图[45]
Figure 2. (a) The mechanism of PDS activation by MnO2 to generate 1O2 [42], (b) The mechanism of PDS activation by the synergistic effect of Co and Mn [23], (c) The mechanism of PMS activation by Fe-doped manganese oxide [44], and (d) The mechanism of PDS activation by Fe-doped manganese oxide [45]
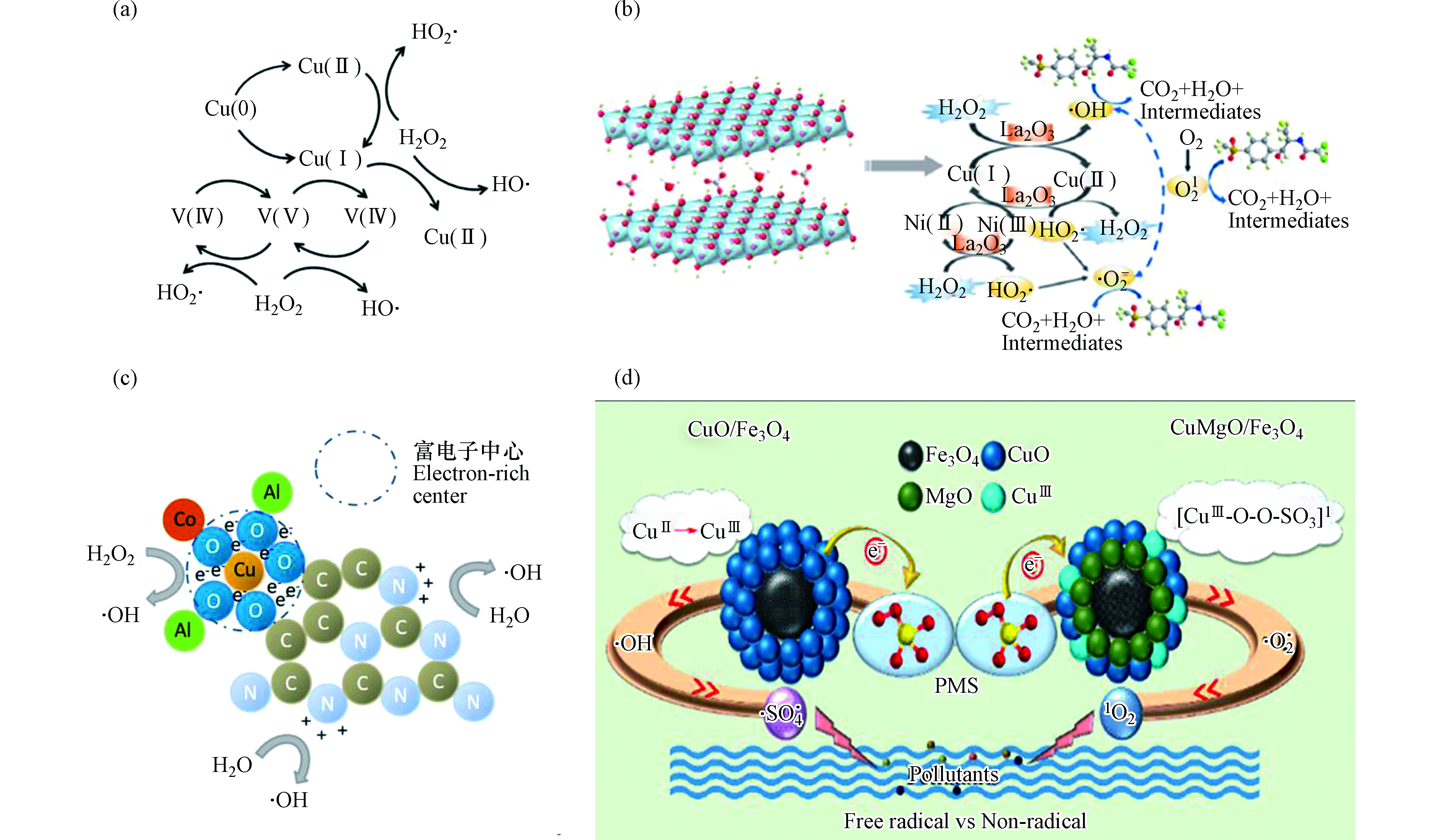
图 4 (a) CuVOx催化过氧化氢类芬顿反应原理[57],(b) CuNiFeLa—LDHs催化过氧化氢类芬顿反应原理[58],(c) 双反应中心催化剂OH—CCN / CuCo—Al2O3活化过氧化氢机理图[61],(d) CuOMgO / Fe3O4的非自由基途径芬顿反应机理图[63]
Figure 4. (a) Schematic illustration of Fenton reactions using CuVOx[57], (b) The mechanism of H2O2 activation by CuNiFeLa—LDHs[58], (c) The mechanism of H2O2 activation by OH—CCN / CuCo—Al2O3 with dual reaction centers[61], and (d) The mechanism Fenton reaction with CuOMgO / Fe3O4 through non-radical pathway[63]

图 5 (a) Al0—CNTs / CNTs-Fe-Cu / O2系统的催化机理[68],(b) Fe2O3 @ FCNT—H芬顿氧化降解污染物示意图[70],(c) FeCo—MCM-41活化PMS反应机理图[75]
Figure 5. (a) The catalytic mechanism of Al0—CNTs / CNTs-Fe-Cu / O2 system[68], (b) The illustration of pollutants degradation in Fe2O3 @ FCNT-H Fenton process [70], and (c) The mechanism PMS activation by FeCo—MCM-41[75]
表 1 催化剂活化H2O2的原理与机制
Table 1. Principle and mechanism of catalysts for the activation of H2O2
反应类型
Reaction type实现途径
Pathway活化过程
Activation process催化材料
Catalysts参考文献
ReferencesH2O2转化为∙OH 增加表面活性位点 产生表面羟基≡Fe—OH,促进H2O2的吸附,H2O2更易于转化为∙OH 天然磁铁矿 [26] 高分散的Fe3O4纳米颗粒,最大化暴露活性位 Fe3O4—NP @ CNF、Fe3O4 MCNs [34] 过渡金属元素的同
构取代钴和锰可形成氧化还原对Co2+ / Co3+和Mn2+ / Mn3+,增加电导率,
并且钴和锰可以增加表面活性位点密度Fe3−xMxO4
(M=Fe, Co, Mn等)[28,31] Fe是活性中心,Co2+ / Co3+氧化还原对可提高催化活性 CoCr2-xFexO4(X=0.0—2.0) [51] 形成中间体 Ru3+与H2O2反应生成中间产物Ru(OOH)2+,Ru3+→ Ru2+,产生·OH RuI3 [76] 协同作用 掺杂金属元素形成双金属催化结构:调节活性金属物种的含量和
表面电荷分布加速·OH的生成Ce5%CuOy [61] 双金属协同促进活性物种生成:Cu+向Cu2+的快速转化加速Fe2+的
再生Fe3O4 / CuO [77] 钒周围形成富电中心和表面氧空位,加快H2O2活化 CuVOx [57] Co2+和Cu+活化H2O2产生·OH,且Cu+可以将生成的Co3+还原为Co2+,实现了Co2+的再生 CuxCo10-x / CNC [59] 形成Fe—O—C结构加速Fe3+ / Fe2+的氧化还原循环 rGS/FexOy / NCL [67] 缺电子与富电子双
反应中心H2O2接受富电子铜中心的电子,形成∙OH;H2O的电子转移到缺电子氮中心,转化为∙OH OH—CCN / CuCo—Al2O3 [61] H2O2转化为多种自由基 协同作用 Fe(III) / Fe(II)、Cu(II) / Cu(I)与Cu(I) / Cu(0)在超声作用下协同将H2O2活化为∙OH和O2·− Cu—Fe3O4 / Cu / C [32] Cu为主要催化活性位点,Ni2+将电子转移至Cu2+,生成更多的Cu+;La2O3可以同时提供Lewis碱性位点和Lewis酸性位点,活化H2O2产生·OH、O2·-和1O2 CuNiFeLa—LDHs [58] Al0—CNTs起活化O2的作用,CNTs—Fe—Cu不仅能活化O2,还可将H2O2催化生成·OH和O2- Al0—CNTs / CNTs—Fe—Cu / O2 [68] 单金属变价 Cu2+被还原为Cu+生成O2·- 3D Cu @ CuO纳米线 [55] Cu2+被还原为Cu+生成O2·-,Cu+通过Cu0再生 Cu2O—Cu / C(CCMs) [54] 形成中间体 Co2+络合物活化H2O2为∙OH和O2·−,或形成中间体直接降解有机
污染物Co(II)络合物 [78]  下载: 导出CSV
下载: 导出CSV
表 2 催化剂活化过硫酸盐的原理与机制
Table 2. Principle and mechanism of catalysts for the activation of persulfate
反应类型
Reaction type实现途径
Pathway活化过程
Activation process催化材料
Catalysts参考文献
References
过硫酸盐生成∙OH和SO4·−单金属变价 通过高价态锰氧化物(Mn4+)还原和低价态锰氧化物(Mn3+)氧化生成SO4-·,SO4-·与H2O的反应生成·OH。 氧化锰八面体分子筛(OMS-2) [40, 79] 超声Cu0转化为Cu+。 nZVC [80- 81] Co2+和Co3+之间单电子转移 。 Co3O4 @ CNT [69] 多金属协同作用 形成≡CoIII / ≡CoII , ≡MnIII / ≡MnII, ≡MnIII / ≡CoII和≡MnIV /≡MnIII等氧化还原电对。 CoxMn3−xO4 [23] Fe—Co协同。 Fe36Co36Si4.8B19.2Nb4 [52] Co—Cu协同。 CoCuAl-LDO [53] Fe—Cu协同。 Fe20Cu80 [82] 形成中间体 亚稳态锰中间体中的超氧离子和自由基氧化或重组产生1O2。 α-MnO2和β-MnO2 [42] 形成表面羟基,与过硫酸根反应生成亚稳态的铜中间体,进而被O2·-氧化生成1O2。 CuOMgO / Fe3O4 [63] 过硫酸盐生成∙OH和SO4·− 强化电子转移 金属掺杂形成氧空位,促进界面电子转移,利于生成∙OH和SO4·−。 HPCMO [45] 过硫酸盐生成∙OH、SO4·−和1O2 多金属协同作用 Co、Mn协同晶格氧和氧空位。 La2CoMnO6−δ [49] Co、Fe协同氧化还原循环活化过硫酸根 FeCo—MCM-41 [75] 形成中间体 Co3O4表面上生成CoOH+,通过氢键与过硫酸根结合,生成SO4∙-。再进一步转化生成·OH和1O2。 纳米立方Co3O4晶体 [50]
下载: 导出CSV
-
[1] BARROS W R P, ERENO T, TAVARES A C, et al. In situ electrochemical generation of hydrogen peroxide in alkaline aqueous solution by using an unmodified gas diffusion electrode [J]. ChemElectroChem, 2015, 2(5): 714-719. doi: 10.1002/celc.201402426 [2] YAN L, HUANG Y Y, CUI J L, et al. Simultaneous As(III) and Cd removal from copper smelting wastewater using granular TiO2 columns [J]. Water Research, 2015, 68: 572-579. doi: 10.1016/j.watres.2014.10.042 [3] LI W, PATTON S, GLEASON J M, et al. UV photolysis of chloramine and persulfate for 1, 4-dioxane removal in reverse-osmosis permeate for potable water reuse [J]. Environmental Science & Technology, 2018, 52(11): 6417-6425. [4] HUA Y N, WANG S, XIAO J, et al. Preparation and characterization of Fe3O4/Gallic acid/graphene oxide magnetic nanocomposites as highly efficient Fenton catalysts [J]. RSC Advances, 2017, 7(46): 28979-28986. doi: 10.1039/C6RA23939K [5] LIU Z, DEMEESTERE K, Van HULLE S. Pretreatment of secondary effluents in view of optimal ozone-based AOP removal of trace organic contaminants: Bench-scale comparison of efficiency and energy consumption [J]. Industrial & Engineering Chemistry Research, 2020, 59(16): 8112-8120. [6] HE D Q, WANG L F, JIANG H, et al. A Fenton-like process for the enhanced activated sludge dewatering [J]. Chemical Engineering Journal, 2015, 272: 128-134. doi: 10.1016/j.cej.2015.03.034 [7] 吕来, 胡春. 多相芬顿催化水处理技术与原理 [J]. 化学进展, 2017, 29(9): 981-999. doi: 10.7536/PC170552 LYU L, HU C. Heterogeneous Fenton catalytic water treatment technology and mechanism [J]. Progress in Chemistry, 2017, 29(9): 981-999(in Chinese). doi: 10.7536/PC170552
[8] XAVIER S, GANDHIMATHI R, NIDHEESH P V, et al. Comparison of homogeneous and heterogeneous Fenton processes for the removal of reactive dye Magenta MB from aqueous solution [J]. Desalination and Water Treatment, 2015, 53(1): 109-118. doi: 10.1080/19443994.2013.844083 [9] CAUDO S, CENTI G, GENOVESE C, et al. Homogeneous versus heterogeneous catalytic reactions to eliminate organics from waste water using H2O2 [J]. Topics in Catalysis, 2006, 40(1/2/3/4): 207-219. [10] LI J Y, PHAM A N, DAI R B, et al. Recent advances in Cu-Fenton systems for the treatment of industrial wastewaters: Role of Cu complexes and Cu composites [J]. Journal of Hazardous Materials, 2020, 392: 122261. doi: 10.1016/j.jhazmat.2020.122261 [11] YAO Y J, CAI Y M, WU G D, et al. Sulfate radicals induced from peroxymonosulfate by cobalt manganese oxides (CoxMn3−xO4) for Fenton-Like reaction in water [J]. Journal of Hazardous Materials, 2015, 296: 128-137. doi: 10.1016/j.jhazmat.2015.04.014 [12] TANG D D, ZHANG G K, GUO S. Efficient activation of peroxymonosulfate by manganese oxide for the degradation of azo dye at ambient condition [J]. Journal of Colloid and Interface Science, 2015, 454: 44-51. doi: 10.1016/j.jcis.2015.05.009 [13] LEE J, Von GUNTEN U, KIM J H. Persulfate-based advanced oxidation: Critical assessment of opportunities and roadblocks [J]. Environmental Science & Technology, 2020, 54(6): 3064-3081. [14] CHENG X, GUO H G, ZHANG Y L, et al. Non-photochemical production of singlet oxygen via activation of persulfate by carbon nanotubes [J]. Water Research, 2017, 113: 80-88. doi: 10.1016/j.watres.2017.02.016 [15] YUN E T, LEE J H, KIM J, et al. Identifying the nonradical mechanism in the peroxymonosulfate activation process: Singlet oxygenation versus mediated electron transfer [J]. Environmental Science & Technology, 2018, 52(12): 7032-7042. [16] LIANG P, ZHANG C, DUAN X G, et al. N-doped graphene from metal-organic frameworks for catalytic oxidation of p-hydroxylbenzoic acid: N-functionality and mechanism [J]. ACS Sustainable Chemistry & Engineering, 2017, 5(3): 2693-2701. [17] LIANG P, ZHANG C, DUAN X G, et al. An insight into metal organic framework derived N-doped graphene for the oxidative degradation of persistent contaminants: Formation mechanism and generation of singlet oxygen from peroxymonosulfate [J]. Environmental Science:Nano, 2017, 4(2): 315-324. doi: 10.1039/C6EN00633G [18] WANG Y B, LIU M, ZHAO X, et al. Insights into heterogeneous catalysis of peroxymonosulfate activation by boron-doped ordered mesoporous carbon [J]. Carbon, 2018, 135: 238-247. doi: 10.1016/j.carbon.2018.01.106 [19] YIN R L, GUO W Q, WANG H Z, et al. Singlet oxygen-dominated peroxydisulfate activation by sludge-derived biochar for sulfamethoxazole degradation through a nonradical oxidation pathway: Performance and mechanism [J]. Chemical Engineering Journal, 2019, 357: 589-599. doi: 10.1016/j.cej.2018.09.184 [20] REN W, NIE G, ZHOU P, et al. The intrinsic nature of persulfate activation and N-doping in carbocatalysis [J]. Environmental Science & Technology, 2020, 54(10): 6438-6447. [21] REN W, XIONG L L, YUAN X H, et al. Activation of peroxydisulfate on carbon nanotubes: Electron-transfer mechanism [J]. Environmental Science & Technology, 2019, 53(24): 14595-14603. [22] SHAO P H, YU S P, DUAN X G, et al. Potential difference driving electron transfer via defective carbon nanotubes toward selective oxidation of organic micropollutants [J]. Environmental Science & Technology, 2020, 54(13): 8464-8472. [23] REN W, XIONG L L, NIE G, et al. Insights into the electron-transfer regime of peroxydisulfate activation on carbon nanotubes: The role of oxygen functional groups [J]. Environmental Science & Technology, 2020, 54(2): 1267-1275. [24] NIDHEESH P V, GANDHIMATHI R, VELMATHI S, et al. Magnetite as a heterogeneous electro Fenton catalyst for the removal of Rhodamine B from aqueous solution [J]. RSC Advances, 2014, 4(11): 5698. doi: 10.1039/c3ra46969g [25] XIA M, LONG M C, YANG Y D, et al. A highly active bimetallic oxides catalyst supported on Al-containing MCM-41 for Fenton oxidation of phenol solution [J]. Applied Catalysis B:Environmental, 2011, 110: 118-125. doi: 10.1016/j.apcatb.2011.08.033 [26] HE H P, ZHONG Y H, LIANG X L, et al. Natural Magnetite: An efficient catalyst for the degradation of organic contaminant [J]. Scientific Reports, 2015, 5(1): 1-10. doi: 10.9734/JSRR/2015/14076 [27] XU L J, WANG J L. Fenton-like degradation of 2, 4-dichlorophenol using Fe3O4 magnetic nanoparticles [J]. Applied Catalysis B:Environmental, 2012, 123/124: 117-126. doi: 10.1016/j.apcatb.2012.04.028 [28] COSTA R C C, LELIS M F F, OLIVEIRA L C A, et al. Novel active heterogeneous Fenton system based on Fe3−xMxO4 (Fe, Co, Mn, Ni): The role of M2+ species on the reactivity towards H2O2 reactions [J]. Journal of Hazardous Materials, 2006, 129(1/2/3): 171-178. [29] MARSAC R, PASTUREL M, HANNA K. Reduction kinetics of nitroaromatic compounds by titanium-substituted magnetite [J]. The Journal of Physical Chemistry C, 2017, 121(21): 11399-11406. doi: 10.1021/acs.jpcc.7b01920 [30] LIANG X L, LI Y, WEI G L, et al. Heterogeneous reduction of 2-chloronitrobenzene by Co-substituted magnetite coupled with aqueous Fe2+: Performance, factors, and mechanism [J]. ACS Earth and Space Chemistry, 2019, 3(5): 728-737. doi: 10.1021/acsearthspacechem.8b00204 [31] LI Y, WEI G L, LIANG X L, et al. Metal substitution-induced reducing capacity of magnetite coupled with aqueous Fe(II) [J]. ACS Earth and Space Chemistry, 2020, 4(6): 905-911. doi: 10.1021/acsearthspacechem.0c00089 [32] XIAO J, LAI J H, LI R C, et al. Enhanced ultrasonic-assisted heterogeneous Fenton degradation of organic pollutants over a new copper magnetite (Cu-Fe3O4/Cu/C) nanohybrid catalyst [J]. Industrial & Engineering Chemistry Research, 2020, 59(27): 12431-12440. [33] HE Z Q, GAO C, QIAN M Q, et al. Electro-Fenton process catalyzed by Fe3O4 magnetic nanoparticles for degradation of C. I. reactive blue 19 in aqueous solution: Operating conditions, influence, and mechanism [J]. Industrial & Engineering Chemistry Research, 2014, 53(9): 3435-3447. [34] WANG W, LIU Y, LI T L, et al. Heterogeneous Fenton catalytic degradation of phenol based on controlled release of magnetic nanoparticles [J]. Chemical Engineering Journal, 2014, 242: 1-9. doi: 10.1016/j.cej.2013.12.080 [35] WANG Y L, ZHU L, YANG X, et al. Facile synthesis of three-dimensional Mn3O4hierarchical microstructures and their application in the degradation of methylene blue [J]. Journal of Materials Chemistry A, 2015, 3(6): 2934-2941. doi: 10.1039/C4TA05493H [36] JO Y H, HONG S H, PARK T J, et al. The synthesized and thermally modified Mn-Ca-FeOOH composite in persulfate system: Its role to discolor methylene blue [J]. Applied Surface Science, 2014, 301: 576-583. doi: 10.1016/j.apsusc.2014.02.134 [37] SAPUTRA E, MUHAMMAD S, SUN H Q, et al. Different crystallographic one-dimensional MnO2 nanomaterials and their superior performance in catalytic phenol degradation [J]. Environmental Science & Technology, 2013, 47(11): 5882-5887. [38] LIU Q R, DUAN X G, SUN H Q, et al. Size-tailored porous spheres of manganese oxides for catalytic oxidation via peroxymonosulfate activation [J]. The Journal of Physical Chemistry C, 2016, 120(30): 16871-16878. doi: 10.1021/acs.jpcc.6b05934 [39] LIU H Z, BRUTON T A, DOYLE F M, et al. In situ chemical oxidation of contaminated groundwater by persulfate: Decomposition by Fe(III)-and Mn(IV)-containing oxides and aquifer materials [J]. Environmental Science & Technology, 2014, 48(17): 10330-10336. [40] TEPE O. Catalytic removal of remazol brilliant blue R by manganese oxide octahedral molecular sieves and persulfate [J]. Journal of Environmental Engineering, 2018, 144(9): 04018087. doi: 10.1061/(ASCE)EE.1943-7870.0001441 [41] TEPE O, TUNÇ Z, YıLDıZ B, et al. Efficient removal of paracetamol by manganese oxide octahedral molecular sieves (OMS-2) and persulfate [J]. Water, Air, & Soil Pollution, 2020, 231(5): 1-15. [42] ZHU S S, LI X J, KANG J, et al. Persulfate activation on crystallographic manganese oxides: Mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants [J]. Environmental Science & Technology, 2019, 53(1): 307-315. [43] ZHU S S, HO S H, JIN C, et al. Nanostructured manganese oxides: Natural/artificial formation and their induced catalysis for wastewater remediation [J]. Environmental Science:Nano, 2020, 7(2): 368-396. doi: 10.1039/C9EN01250H [44] YU L, ZHANG G, LIU C L, et al. Interface stabilization of undercoordinated iron centers on manganese oxides for nature-inspired peroxide activation [J]. ACS Catalysis, 2018, 8(2): 1090-1096. doi: 10.1021/acscatal.7b03338 [45] TIAN N, TIAN X K, NIE Y L, et al. Enhanced 2, 4-dichlorophenol degradation at pH 3-11 by peroxymonosulfate via controlling the reactive oxygen species over Ce substituted 3D Mn2O3 [J]. Chemical Engineering Journal, 2019, 355: 448-456. doi: 10.1016/j.cej.2018.08.183 [46] CHEN X Y, CHEN J W, QIAO X L, et al. Performance of nano-Co3O4/peroxymonosulfate system: Kinetics and mechanism study using Acid Orange 7 as a model compound [J]. Applied Catalysis B:Environmental, 2008, 80(1/2): 116-121. [47] ANIPSITAKIS G P, DIONYSIOU D D. Radical generation by the interaction of transition metals with common oxidants [J]. Environmental Science & Technology, 2004, 38(13): 3705-3712. [48] LIN K Y A, CHEN B J, CHEN C K. Evaluating Prussian blue analogues MII3[MIII(CN)6]2 (MII = Co, Cu, Fe, Mn, Ni;MIII = Co, Fe) as activators for peroxymonosulfate in water [J]. RSC Advances, 2016, 6(95): 92923-92933. doi: 10.1039/C6RA16011E [49] LUO X S, BAI L M, XING J J, et al. Ordered mesoporous cobalt containing perovskite as a high-performance heterogeneous catalyst in activation of peroxymonosulfate [J]. ACS Applied Materials & Interfaces, 2019, 11(39): 35720-35728. [50] YANG W C, LI X Y, JIANG Z, et al. Structure-dependent catalysis of Co3O4 crystals in persulfate activation via nonradical pathway [J]. Applied Surface Science, 2020, 525: 146482. doi: 10.1016/j.apsusc.2020.146482 [51] TATARCHUK T, SHYICHUK A, TRAWCZYŃSKA I, et al. Spinel cobalt(II) ferrite-chromites as catalysts for H2O2 decomposition: Synthesis, morphology, cation distribution and antistructure model of active centers formation [J]. Ceramics International, 2020, 46(17): 27517-27530. doi: 10.1016/j.ceramint.2020.07.243 [52] JIANG J L, JIA Z, HE Q, et al. Synergistic function of iron and cobalt in metallic glasses for highly improving persulfate activation in water treatment [J]. Journal of Alloys and Compounds, 2020, 822: 153574. doi: 10.1016/j.jallcom.2019.153574 [53] LUO L, WANG Y L, ZHU M L, et al. Co-Cu-Al layered double oxides as heterogeneous catalyst for enhanced degradation of organic pollutants in wastewater by activating peroxymonosulfate: Performance and synergistic effect [J]. Industrial & Engineering Chemistry Research, 2019, 58(20): 8699-8711. [54] SUN B F, LI H L, LI X Y, et al. Degradation of organic dyes over Fenton-like Cu2O-Cu/C catalysts [J]. Industrial & Engineering Chemistry Research, 2018, 57(42): 14011-14021. [55] SU Z, LI J, ZHANG D D, et al. Novel flexible Fenton-like catalyst: Unique CuO nanowires arrays on copper mesh with high efficiency across a wide pH range [J]. Science of the Total Environment, 2019, 647: 587-596. doi: 10.1016/j.scitotenv.2018.08.022 [56] ZHANG N Q, YI Y Q, LIAN J T, et al. Effects of Ce doping on the Fenton-like reactivity of Cu-based catalyst to the fluconazole [J]. Chemical Engineering Journal, 2020, 395: 124897. doi: 10.1016/j.cej.2020.124897 [57] ZHANG N Q, XUE C J, WANG K, et al. Efficient oxidative degradation of fluconazole by a heterogeneous Fenton process with Cu-V bimetallic catalysts [J]. Chemical Engineering Journal, 2020, 380: 122516. doi: 10.1016/j.cej.2019.122516 [58] CHEN T, ZHU Z L, ZHANG H, et al. Enhanced removal of veterinary antibiotic florfenicol by a Cu-based Fenton-like catalyst with wide pH adaptability and high efficiency [J]. ACS Omega, 2019, 4(1): 1982-1994. doi: 10.1021/acsomega.8b03406 [59] WANG J, LIU C, FENG J Y, et al. MOFs derived Co/Cu bimetallic nanoparticles embedded in graphitized carbon nanocubes as efficient Fenton catalysts [J]. Journal of Hazardous Materials, 2020, 394: 122567. doi: 10.1016/j.jhazmat.2020.122567 [60] BOKARE A D, CHOI W. Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes [J]. Journal of Hazardous Materials, 2014, 275: 121-135. doi: 10.1016/j.jhazmat.2014.04.054 [61] LYU L, ZHANG L L, HE G Z, et al. Selective H2O2 conversion to hydroxyl radicals in the electron-rich area of hydroxylated C-g-C3N4/CuCo–Al2O3 [J]. Journal of Materials Chemistry A, 2017, 5(15): 7153-7164. doi: 10.1039/C7TA01583F [62] LEI Y, CHEN C S, TU Y J, et al. Heterogeneous degradation of organic pollutants by persulfate activated by CuO-Fe3O4: Mechanism, stability, and effects of pH and bicarbonate ions [J]. Environmental Science & Technology, 2015, 49(11): 6838-6845. [63] JAWAD A, ZHAN K, WANG H B, et al. Tuning of persulfate activation from a free radical to a nonradical pathway through the incorporation of non-redox magnesium oxide [J]. Environmental Science & Technology, 2020, 54(4): 2476-2488. [64] BELLO M M, RAMAN A A A, ASGHAR A. Activated carbon as carrier in fluidized bed reactor for Fenton oxidation of recalcitrant dye: Oxidation-adsorption synergy and surface interaction [J]. Journal of Water Process Engineering, 2020, 33: 101001. doi: 10.1016/j.jwpe.2019.101001 [65] BOUNAB L, IGLESIAS O, GONZÁLEZ-ROMERO E, et al. Effective heterogeneous electro-Fenton process of m-cresol with iron loaded actived carbon [J]. RSC Advances, 2015, 5(39): 31049-31056. doi: 10.1039/C5RA03050A [66] WANG Y J, ZHAO G H, CHAI S N, et al. Three-dimensional homogeneous ferrite-carbon aerogel: One pot fabrication and enhanced electro-Fenton reactivity [J]. ACS Applied Materials & Interfaces, 2013, 5(3): 842-852. [67] YAO T J, JIA W J, FENG Y, et al. Preparation of reduced graphene oxide nanosheet/FexOy/nitrogen-doped carbon layer aerogel as photo-Fenton catalyst with enhanced degradation activity and reusability [J]. Journal of Hazardous Materials, 2019, 362: 62-71. doi: 10.1016/j.jhazmat.2018.08.084 [68] CHEN Y, YANG Z, LIU Y B, et al. Fenton-like degradation of sulfamerazine at nearly neutral pH using Fe-Cu-CNTs and Al0-CNTs for in situ generation of H2O2/OH/O2− [J]. Chemical Engineering Journal, 2020, 396: 125329. doi: 10.1016/j.cej.2020.125329 [69] LIU B M, SONG W B, WU H X, et al. Degradation of norfloxacin with peroxymonosulfate activated by nanoconfinement Co3O4@CNT nanocomposite [J]. Chemical Engineering Journal, 2020, 398: 125498. doi: 10.1016/j.cej.2020.125498 [70] YANG Z C, QIAN J S, YU A Q, et al. Singlet oxygen mediated iron-based Fenton-like catalysis under nanoconfinement [J]. PNAS, 2019, 116(14): 6659-6664. doi: 10.1073/pnas.1819382116 [71] BENZAQUÉN T B, OCHOA RODRIGUEZ P A, CÁNEPA A L, et al. Heterogeneous Fenton reaction for the treatment of ACE in residual waters of pharmacological origin using Fe-SBA-15 nanocomposites [J]. Molecular Catalysis, 2020, 481: 110239. doi: 10.1016/j.mcat.2018.11.010 [72] BENZAQUÉN T B, BARRERA D A, CARRARO P M, et al. Nanostructured catalysts applied to degrade atrazine in aqueous phase by heterogeneous photo-Fenton process [J]. Environmental Science and Pollution Research, 2019, 26(5): 4192-4201. doi: 10.1007/s11356-018-2348-9 [73] MAZILU I, CIOTONEA C, CHIRIEAC A, et al. Synthesis of highly dispersed iron species within mesoporous (Al-)SBA-15 silica as efficient heterogeneous Fenton-type catalysts [J]. Microporous and Mesoporous Materials, 2017, 241: 326-337. doi: 10.1016/j.micromeso.2016.12.024 [74] YIN Y, SHI L, LI W L, et al. Boosting Fenton-like reactions via single atom Fe catalysis [J]. Environmental Science & Technology, 2019, 53(19): 11391-11400. [75] SUN X W, XU D Y, DAI P, et al. Efficient degradation of methyl orange in water via both radical and non-radical pathways using Fe-Co bimetal-doped MCM-41 as peroxymonosulfate activator [J]. Chemical Engineering Journal, 2020, 402: 125881. doi: 10.1016/j.cej.2020.125881 [76] ROKHINA E V, LAHTINEN M, NOLTE M C M, et al. The influence of ultrasound on the RuI3-catalyzed oxidation of phenol: Catalyst study and experimental design [J]. Applied Catalysis B:Environmental, 2009, 87(3/4): 162-170. [77] GHASEMI H, AGHABARARI B, ALIZADEH M, et al. High efficiency decolorization of wastewater by Fenton catalyst: Magnetic iron-copper hybrid oxides [J]. Journal of Water Process Engineering, 2020, 37: 101540. doi: 10.1016/j.jwpe.2020.101540 [78] LÁZARO-MARTÍNEZ J M, LOMBARDO LUPANO L V, PIEHL L L, et al. New insights about the selectivity in the activation of hydrogen peroxide by cobalt or copper hydrogel heterogeneous catalysts in the generation of reactive oxygen species [J]. The Journal of Physical Chemistry C, 2016, 120(51): 29332-29347. doi: 10.1021/acs.jpcc.6b10957 [79] ZHANG H, WU J, WANG Z Q, et al. Electrochemical oxidation of Crystal Violet in the presence of hydrogen peroxide [J]. Journal of Chemical Technology & Biotechnology, 2010, 85(11): 1436-1444. [80] ZHANG T T, YANG Y L, LI X, et al. Degradation of sulfamethazine by persulfate activated with nanosized zero-valent copper in combination with ultrasonic irradiation [J]. Separation and Purification Technology, 2020, 239: 116537. doi: 10.1016/j.seppur.2020.116537 [81] WANG Q, CAO Y, ZENG H, et al. Ultrasound-enhanced zero-valent copper activation of persulfate for the degradation of bisphenol AF [J]. Chemical Engineering Journal, 2019, 378: 122143. doi: 10.1016/j.cej.2019.122143 [82] MANICKAM-PERIYARAMAN P, ESPINOSA J C, FERRER B, et al. Bimetallic iron-copper oxide nanoparticles supported on nanometric diamond as efficient and stable sunlight-assisted Fenton photocatalyst [J]. Chemical Engineering Journal, 2020, 393: 124770. doi: 10.1016/j.cej.2020.124770 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 30879
- HTML全文浏览数: 30879
- PDF下载数: 1458
- 施引文献: 0
