

-
氨气(NH3)是大气中重要的碱性气体,参与氮的循环过程,在二次无机气溶胶的形成中起重要作用,是地区大气复合污染的重要前体物之一[1 − 6]. NH3可与大气中的NOx或SO2反应生成硝酸铵盐或硫酸铵盐颗粒,在气溶胶初始核化过程中扮演着极其重要的角色,是大气雾霾污染过程中二次颗粒物形成的重要因素[7 − 9]. 《中华人民共和国环境保护税法》法案规定NH3为应税污染物. NH3不仅对雾霾形成有重要贡献,还会通过大气氮素沉降造成土壤和地表水酸化、水体富营养化,干扰生态系统中的氮素平衡. 中国农业大学刘学教授军曾在《Nature》发文分析中国氮素沉降对大气污染的影响,并指出NH3是我国氮素污染的主要来源[10]. 此外,NH3具有强烈的刺激性气味,对人体皮肤组织和呼吸道有刺激作用. 由于人类具有嗅觉疲劳,若长时间接触低浓度NH3,可能难以察觉,对人体健康有十分严重的影响[11 − 13]. NH3的嗅觉阈值为1.04 mg·m−3[14],美国职业安全与健康管理局设定了NH3的短期(15 min)接触极限为24.3 mg·m−3,长期暴露于浓度超过34.8 mg·m−3的NH3可导致暂时失明、肺部疾病、烧伤和水泡,甚至有生命危险[15]. NH3排放的最大来源是农业,包括畜牧业和氮肥的施用[16],其次是工业过程、车辆排放以及土壤和海洋的挥发[17]. 在室内环境中,NH3主要产生于建筑装修材料和人体代谢释放[18]. 建筑材料混凝土中含有尿素防冻剂、装修材料和家具中含有氨水成分添加剂等都会缓慢释放NH3. 此外,NH3也是人体释放的主要污染物之一. 研究发现,成年人在25 °C、27 °C和29 °C时,NH3释放速率可分别达到每人0.4 mg·h−1、0.8 mg·h−1和1.4 mg·h−1[19]. 因此,如何控制氨排放和高效去除NH3,对提高大气环境质量和保障人体健康具有重要的现实意义和实际需求.
目前常用的除氨方法有热焚烧法、冷凝法、生物过滤法、选择性催化氧化法、光催化氧化法、吸附法等[20-21]. 其中,吸附法由于操作简便,高效节能,稳定性高,是工程实践中最常用的方法,也是当前最常用的深度除氨方法之一. 显然,NH3吸附净化的关键在于高性能的吸附剂. 因此,开发吸附容量大、吸附速率快、选择性高、易再生的吸附剂是实现NH3深度去除的关键. 在工程实践中,活性炭、沸石、膨润土、金属无机化合物等多孔材料是目前常用的NH3吸附材料[22]. 未经改性的颗粒活性炭(GAC)和粉末活性炭(PAC)对NH3的吸附性能一般较低[23 − 26]. Takahashi等[26]研究发现13X沸石和离子交换树脂对痕量NH3有一定的吸附能力,但吸附容量有限. Rezaei等[27]评价了MgO、ZnO、TiO2等金属氧化物纳米颗粒NH3吸附性能,发现其中TiO2的吸附容量最高. 一些复合材料和改性材料也被开发用于NH3的吸附,例如活性炭AC负载TiO2的TiO2-AC复合材料[28],碳纤维复合材料(ACFCs)[29]等,均展现出不错的吸附性能. 此外,Zhang[30]和Grant[31]等还制备并评价了一系列金属有机框架材料(MOFs)的NH3吸附性能,结果表明,MOFs的氨气吸附性能明显高于其他类材料. 然而,尽管它们在NH3去除方面表现出优异的性能,但与其他商业化产品相比,MOFs材料较高的生产成本严重限制了其大规模实际应用.
多孔吸附材料通常存在的主要问题是吸附容量小和再生困难. 由于吸附容量不高,就需要频繁的再生或更换吸附剂. 目前,除活性炭沸石等吸附材料外,过渡金属氧化物类吸附剂引起部分研究者的关注[32 − 34]. 与此同时,本课题组着眼于目前研究较少的无载体过渡金属氧化物自身NH3吸附性能,发现二氧化锰(MnO2)纳米材料具有NH3吸附潜力,并对比研究了一系列不同隧道结构的MnO2(β-MnO2(1×1)、α-MnO2(2×2)、γ-MnO2(1×2)、δ-MnO2(1×∞)、τ-MnO2(3×3))对NH3的吸附再生性能. 研究发现,MnO2的隧道尺寸对NH3吸附性能影响显著. 其中,具有2×2隧道结构的α-MnO2吸附性能最好[35]. 通常认为,吸附剂比表面积显著影响物理吸附性能,而吸附剂的微观形貌显著影响其比表面积. 此外,NH3作为碱性气体易于吸附在材料的表面酸性位点上. 前期研究[35]表明α-MnO2优异的NH3吸附性能主要取决于两方面:一是,α-MnO2的隧道尺寸和NH3空气动力学直径相近(约0.44 nm),有利于NH3的物理吸附;二是,α-MnO2具有较多较强的表面酸性位点,尤其是Brønsted酸性位,大幅增加了对碱性气体NH3的化学吸附. 因此,在α-MnO2基础之上,通过调控制备有利于NH3吸附的微观形貌和强化表面酸性位,从而同时增强NH3的物理吸附和化学吸附,有望能够显著改善NH3吸附性能,有助于实现氨的减排控制.
本文在前期锰氧化物氨气吸附材料研究基础之上,选择α-MnO2开展进一步的优化深入研究,合成了一系列不同形貌的α-MnO2纳米材料(纳米线、纳米管、纳米棒、纳米花),并对其进行表面酸性位强化,以期研制具有优异吸附性能的NH3吸附材料. 此外,探究吸附温度、初始NH3浓度、吸附剂用量对NH3吸附性能的影响. 最后,结合材料表征和原位红外等,进一步分析隧道结构、吸附位点和吸附机制,揭示α-MnO2形貌和表面酸性位在NH3吸附中的作用.
-
α-MnO2纳米线的制备[36]:α-MnO2纳米线是通过KMnO4和(NH4)2C2O4·H2O之间的水热氧化还原反应制备. 具体而言,将
3.9509 g KMnO4和1.4211 g (NH4)2C2O4·H2O溶解在70 mL去离子水中. 在强烈的磁性搅拌下搅拌约30 min,然后将混合均匀的溶液转移到100 mL水热反应釜中. 水热反应釜在烘箱内以180 ℃加热24 h. 反应釜自然冷却至室温后,通过过滤收集最终产物,用去离子水洗涤数次,并在105 ℃下干燥.α-MnO2纳米管的制备[37]:将
0.6080 g KMnO4溶解在70 mL去离子水中,加入1.27 mL质量分数为37% HCl溶液. 然后将混合均匀的溶液转移到100 mL水热反应釜中. 水热反应釜在烘箱内以140 ℃加热12 h. 反应釜自然冷却至室温后,过滤收集最终产物,去离子水洗涤数次,并在80 ℃下干燥.α-MnO2纳米棒的制备[38]:将
0.9482 g KMnO4和1.5211 g MnSO4·H2O溶解在60 mL去离子水中,加入3 mL 质量分数为98% H2SO4溶液. 然后将混合均匀的溶液转移到100 mL水热反应釜中. 水热反应釜在烘箱内以160 ℃加热12 h. 反应釜自然冷却至室温后,过滤收集最终产物,用去离子水洗涤数次,并在105 ℃下干燥.α-MnO2纳米花的制备[38]:将
2.1626 g K2S2O8,1.3521 g MnSO4·H2O,1.3941 g K2SO4分别溶解在60 mL去离子水中,加入2 mL质量分数为98% H2SO4溶液. 然后将混合均匀的溶液转移到100 mL水热反应釜中. 水热反应釜在烘箱内以140 ℃加热12 h. 反应釜自然冷却至室温后,过滤收集最终产物,用去离子水洗涤数次,并在105 ℃下干燥.表面酸性位强化不同形貌α-MnO2的制备:以表面酸性位强化α-MnO2纳米线制备过程为例,将制备好的0.5 g α-MnO2纳米线粉末浸入50 mL 0.5 mol·L−1硝酸溶液中,在室温下搅拌12 h. 搅拌完成后,将悬浮液过滤,用去离子水反复清洗,直到洗脱液呈近中性. 随后将得到的固体在105 ℃下干燥. 得到的表面酸性位强化α-MnO2纳米线记为纳米线-H. 同样,根据上述操作步骤制备纳米管-H、纳米棒-H、纳米花-H.
碳材料的制备:用于NH3吸附性能对比的碳基材料均为购买的商品材料. 商业椰壳活性炭购自深圳兴万邦活性炭有限公司. 商业煤基活性炭和竹炭购自卡尔冈炭素(天津)有限公司. 表面酸性位强化碳材料的制备方法与表面酸性位强化α-MnO2相同,分别记为椰壳活性炭-H、煤基活性炭-H、竹炭-H.
-
采用X射线衍射仪(Bruker D8-Advance XRD, Germany)对表面酸性位强化前后不同形貌α-MnO2吸附剂进行X射线衍射分析,使用Cu Kα辐射源,扫描速度为10(°)·min−1;采用Regulus
8100 场发射电子显微镜(Hitachi, Japan)拍摄α-MnO2吸附剂的扫描电子显微镜照片(FESEM);采用MicromeriticsASAP2460 物理吸附分析仪(Micromeritics, USA)测定表面酸性位强化前后不同形貌α-MnO2吸附剂的比表面积、孔隙体积和平均孔径,使用Brunauer−Emmett−Teller(BET)模型计算样品的比表面积;采用Thermo Scientific K-Alpha+X射线光电子能谱仪(XPS, Thermo, USA)进行表面酸性位强化前后不同形貌α-MnO2吸附剂的X射线光电子能谱分析;采用Micromeritics AutoChem II2920 化学吸附仪(Micromeritics, USA)进行氨气程序升温脱附(NH3-TPD)测试;采用Thermo Scientific iS50原位红外光谱仪(in-situ DRIFTS, Thermo, USA)检测分析NH3在表面酸性位强化前后不同形貌α-MnO2吸附剂上吸附过程的傅里叶变换红外光谱. -
吸附剂性能评价实验在一套连续流固定床反应器内完成. 将吸附剂(100 mg,40—60目)装载在石英管固定床反应器中,通过动态穿透实验研究不同吸附剂对NH3的吸附性能. 配气系统分为干气和NH3,以合成空气(21% O2,79% N2)作为载气,将合成空气流过置于恒温水浴(5 ℃)中玻璃瓶内的氨水溶液,以此产生气态NH3. 通过数字式质量流量计控制调节混合气体总流量为100 mL·min−1,对应的气体空速(GHSV)为60 L·g−1·h−1. 通过傅里叶变换红外多组分气体分析仪(Thermo Scientific Antaris IGS, USA)检测NH3的进口和出口浓度. 通过对穿透曲线进行积分得到吸附剂对NH3的吸附容量(mg NH3·g−1吸附剂). 单位质量吸附剂的平衡吸附容量通过如下公式(1)计算:
式中,qe为吸附容量(mg·g−1),C0和Ct分别为进口和出口NH3浓度(mg·L−1),V为气体流速(L·min−1),t为吸附饱和时间(min),m为吸附剂质量(g).
-
本研究通过控制反应前驱体和水热条件,合成了纳米线、纳米管、纳米棒和纳米花等4种形貌的α-MnO2,并通过酸浸渍调节了表面酸性位点. 首先,采用XRD对酸性位调控前后4种不同形貌的α-MnO2的晶体结构进行了表征. 如图1所示,所有样品都具有类似的衍射峰,可以归属于标准的隐钾锰矿型MnO2(α-MnO2, JCPDS, PDF #44-0141),表明了α-MnO2的成功合成. 此外,可以发现制备的样品均为纯相α-MnO2,除α-MnO2的衍射峰外,没有观察到其他的衍射杂峰. α-MnO2由[MnO6]八面体通过共角和共边构建而成,具有一种结构明确的[2×2]隧道结构,隧道尺寸约为0.46 nm[39]. 另外,对比酸性位调控前后α-MnO2的XRD,可以发现通过简单酸浸法来增强α-MnO2表面的酸性位点,对其晶体结构没有明显的影响.
-
图2展示了酸性位调控前后α-MnO2的不同形貌结构. 可以发现,不同反应前驱体和水热条件制备的α-MnO2具有明显不同的形貌. 高锰酸钾和草酸铵在180 ℃水热反应的最终产物为均匀纳米线形态(图2a);高锰酸钾与盐酸在140 ℃下水热反应制备的样品呈现中空纳米管形态(图2c);高锰酸钾和硫酸锰在160 ℃下水热反应得到的最终产物为实心纳米棒形态(图2e);过硫酸钾和硫酸锰在140 ℃下水热氧化还原反应得到的样品为纳米棒组成的球状纳米花形态(图2g). 另外,可以观察到酸性位调控前后两组α-MnO2的形态基本一致,表明α-MnO2表面酸性位的强化没有改变其表面形貌结构.
-
在室温下通过动态穿透实验,对比研究了不同形貌结构α-MnO2的NH3吸附性能. 结果如图3所示,不同α-MnO2样品穿透时间和吸附能力顺序为:纳米线-H>纳米棒-H>纳米棒>纳米花-H>纳米花>纳米管>纳米管-H>纳米线,其NH3吸附容量依次为12.06、7.32、7.05、6.56、5.95、5.10、4.94、0.15 mg·g−1. 由图3a可见,随着吸附容量的上升,NH3吸附穿透曲线逐渐右移. 特别是,酸性位增强后的α-MnO2纳米线具有最佳的NH3吸附性能,其吸附容量达到12.06 mg·g−1. 对比酸性位增强前后,纳米管、纳米棒和纳米花的吸附容量没有显著变化,但纳米线的NH3吸附容量却从0.15 mg·g−1剧增到了12.06 mg·g−1,提升了80倍以上. 上述结果表明,α-MnO2的4种形貌结构中纳米线的表面酸性位更容易被调控和强化,也说明了表面酸性位的强化可以显著提高NH3的吸附性能.
-
对于吸附性能最好的α-MnO2纳米线-H,进一步探究了吸附温度、初始NH3浓度和吸附剂用量对其NH3吸附性能的影响. 吸附温度对纳米线-H的NH3吸附穿透曲线及吸附容量的影响如图4a、b所示. 当温度升高时,NH3的穿透曲线逐渐向左移动,表明NH3的吸附性能下降. 这是因为NH3在α-MnO2上的吸附是放热过程[27,40],高温会增强吸附质的解吸,降低吸附容量. 另一方面,温度升高会加剧NH3分子的热运动,动能随之变大,导致吸附位点更难捕获NH3分子,最终导致吸附性能下降[41]. 图4c、d显示了NH3初始浓度对NH3穿透曲线和吸附能力的影响. 可以发现,穿透曲线随着NH3浓度的增加明显向左移动,说明NH3穿透加快. 但初始NH3浓度对吸附容量的影响较小,不同浓度NH3下的吸附容量非常接近. 高初始NH3浓度时吸附容量略大,这可能是由于浓度增加引起传质推动力增大所致. 此外,还研究了吸附剂使用量对NH3吸附性能的影响. 如图4e、f所示,在实验的3种不同吸附剂用量下NH3的平均吸附容量为13.31 mg·g−1,接近初始值12.06 mg·g−1. 由此可见,NH3平衡吸附容量基本不受吸附剂使用量影响. 在吸附剂使用量较少(50 mg)时吸附容量略大,这可能是由于吸附剂用量减少使得吸附剂在固定式填充床中的床层高度降低,内部传质阻力减小,吸附速率提升所致[42].
-
在NH3净化的实际工程应用中,以活性炭为代表的各种碳基多孔材料是最常用的吸附剂. 因此,将α-MnO2纳米线-H的吸附性能与煤基活性炭、椰壳活性炭、竹炭3种应用广泛的商业碳基吸附剂进行了对比. 由图5可见,α-MnO2纳米线-H表现出最佳的吸附性能,明显优于其他碳基材料及其相应的酸性强化材料. 与未经酸性位强化的商业碳基材料相比,α-MnO2纳米线-H的NH3吸附能力提高了6—12倍,进一步证实了α-MnO2纳米线-H是一种具有实际应用潜力的NH3吸附材料.
-
鉴于α-MnO2纳米线-H对NH3的优异吸附性能,探究NH3吸附机理以及吸附性能与表面结构性质的关系十分有必要. 理论上,气态NH3在吸附剂上的吸附包括物理吸附和化学吸附两个过程. 其中,物理吸附与吸附剂的孔隙结构和比表面积直接相关. 因此,测试了表面酸性位强化后不同形貌结构α-MnO2样品的比表面积,相关数据汇总如表1所示. 图6为4种不同形貌结构的α-MnO2及其酸性位强化后样品的N2吸附-脱附等温线. 可以发现,所有α-MnO2样品的等温线均为IUPAC分类标准中IV型等温线,且具有H3型回滞环,这表明它们是具有不均匀孔道的介孔材料[43]. 这可以从图6b和表1中的孔径分布数据得到验证. 此外,结果还表明,酸性位强化前α-MnO2纳米线具有最大比表面积,为88.21 m2·g−1,其次为纳米棒、纳米管和纳米花;而酸性位强化后比表面积大小顺序未发生变化,依旧是纳米线-H>纳米棒-H>纳米管-H>纳米花-H. 其中纳米线经酸处理后比表面积显著增大,达到了126.86 m2·g−1,而其他三种形貌的样品比表面积只略微增加. 众所周知,增加吸附剂的比表面积有利于提高其对吸附质的物理吸附. 这也是α-MnO2纳米线经过酸化处理后,NH3吸附性能得到大幅度提高的原因之一. 但另一方面,结合各样品的比表面积和吸附容量(图3b)可以发现,纳米线具有较大的比表面积,但它的吸附容量却是最小的,另外比表面积的大小顺序与吸附容量的大小顺序并不一一对应. 上述结果也表明,比表面积是影响NH3吸附性能的重要因素,但并不是决定性因素.
-
通过XPS表征对比分析了未处理纳米线和表面酸性位强化后纳米线-H的表面化学组成和元素价态信息. 根据图7a中的Mn 3s的结合能差计算了Mn的平均氧化态(AOS). 表2中结果显示,在酸浸渍后,Mn的AOS有降低的趋势. AOS的降低表明Mn4+含量的下降,为了保持电荷平衡,MnO2中的晶格氧会相应减少,从而产生氧空位[43]. 这意味着纳米线-H上形成了更多的氧空位. Liu[44]和Zhou[45]研究也报道了类似现象,即酸处理会降低锰氧化物的AOS值. 如图7b所示,O 1s谱中拟合出了表面晶格氧(Olatt)、表面吸附氧(Oads)和表面吸附水(Owater)三个峰. 由图7c和表2可知,经酸性位强化的纳米线-H的Oads/Olatt比例高于未处理纳米线,说明纳米线-H具有更丰富表面吸附氧. 此结果与前文提到的氧空位增加相一致. 因为氧分子通常易于吸附在氧化物材料的氧空位上,进而活化成为表面吸附氧/活性氧[46]. 此外,这些氧空位也可能会被水或其解离物占据(即表面羧基),成为潜在的Brønsted酸性位点[44]. 另外,根据图7d和表2中K/Mn变化可以发现,酸浸渍明显降低了α-MnO2中K+的含量,这是由酸浸过程中α-MnO2隧道结构中填充的K+和酸液中H+之间的离子交换造成的.
-
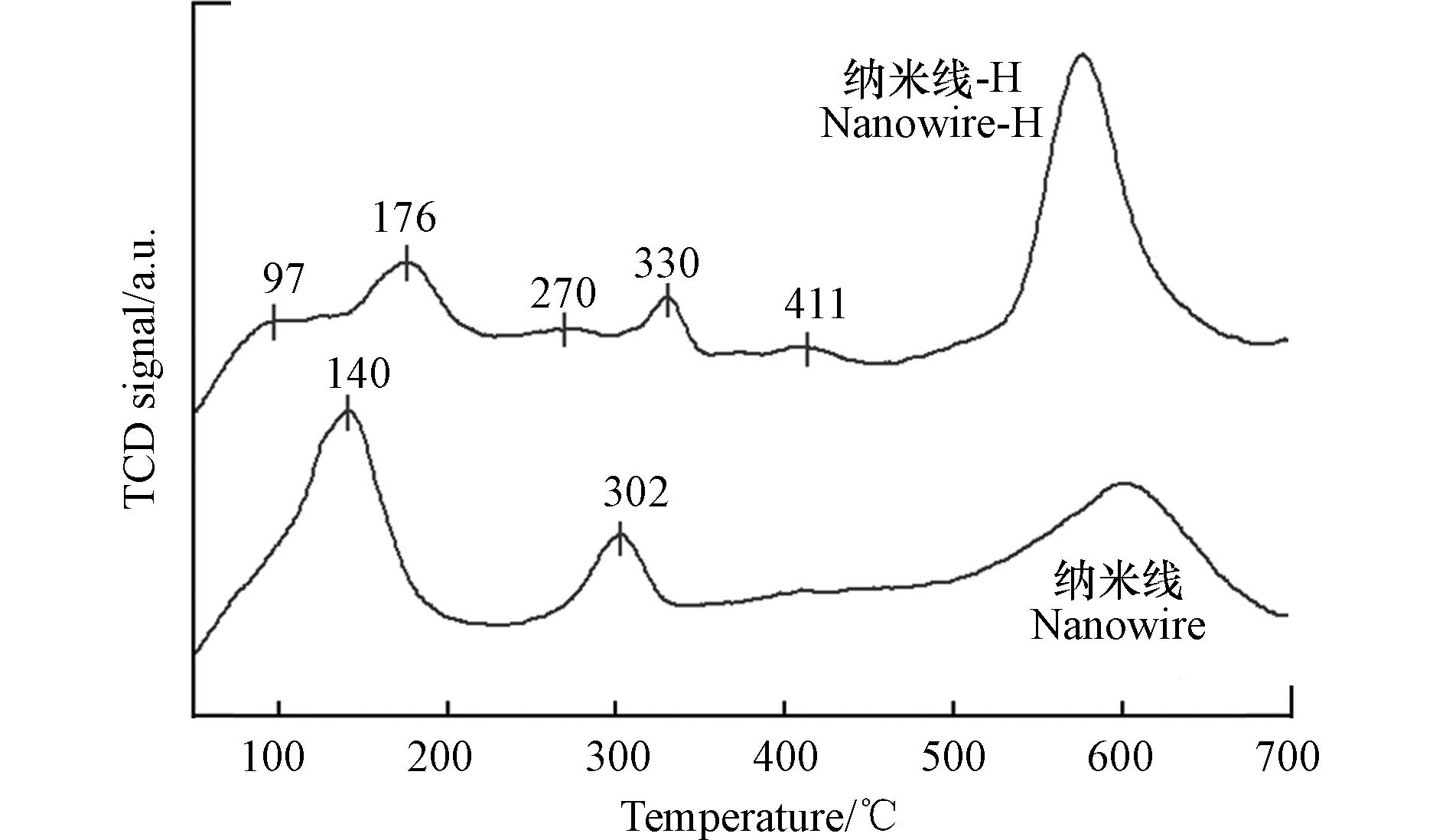
众所周知,NH3作为一种碱性气体,很容易与吸附剂表面的酸性位点结合. 酸性位的数量、强度和性质与其NH3吸附能力密切相关. 一般来说,NH3与吸附剂表面酸性位主要有两种结合方式[47]:一是氧化物的金属阳离子接收氮原子的孤对电子,在表面充当Lewis酸位点;二是NH3被表面含氧官能团或水的质子质子化,然后通过与Brønsted酸位点的酸碱相互作用吸附在表面. 因此,为研究NH3在吸附剂表面酸性位上的吸附情况,采用了NH3-TPD对其表面酸性位性质进行分析. 一般而言,吸附在Lewis酸性位上的NH3倾向于弱吸附能力,会优先脱附;而吸附在Brønsted酸性位上的NH3具有较强的吸附能力,往往滞后脱附. 相关研究表明[48 − 49],一般低温(低于300 ℃)的解吸峰可以归结为与Lewis酸性位结合的NH3;而高温(高于300 ℃)的峰可以归结为吸附在Brønsted酸性位上NH3的解吸. 值得注意的是,锰氧化物在高温下会解吸出大量的晶格氧,干扰TCD信号,而500 ℃以上的峰主要是晶格氧解吸产生的,因此分析时只需考虑500 ℃以前的解吸峰. 如图8所示,α-MnO2纳米线在140 ℃和302 ℃处各存在一个解吸峰,前者可归结为与Lewis酸性位结合的NH3的解吸,后者可归结为吸附在Brønsted酸性位上的NH3的解吸. 对比表面酸性位强化前后的变化,可以发现纳米线-H增加了97、270、411 ℃三处解吸峰,表明NH3吸附酸性位点增多. 此外,原来的两个解吸峰的峰位向右移动了30 ℃左右,表明酸性位强度的增强. 以上结果证明了酸浸渍可以极大地增强α-MnO2纳米线中酸性位点的数量和强度. 显然,更多更强的表面酸性位会更有利于NH3的化学吸附,这也是酸性位增强后纳米线-H吸附性能显著提升的主要原因.
-
原位红外(in-situ DRIFTS)技术在研究材料微观结构和反应活性以及探讨反应机理方面有着其它技术无法比拟的优越性,特别适合于固体粉末样品的表面结构和表面吸附物种的鉴别[50 − 51]. 为了进一步研究α-MnO2吸附剂对NH3的吸附机理,采用原位红外对α-MnO2纳米线和纳米线-H在不同时间暴露于NH3后的表面物种进行分析. 由图9a可知,与新鲜α-MnO2纳米线相比,NH3吸附后纳米线表面在
1541 、1454 、1388 cm−1处出现新的吸收峰,且随着暴露时间的延长,峰强度均有一定程度的增加直至稳定. 其中1541 cm−1处的峰为NH3吸附在Lewis酸位上的特征峰,而1454 cm−1和1388 cm−1处的峰与化学吸附在Brønsted酸性位上的NH4+的振动频率相近[52 − 53]. 此外,3000 —3500 cm−1处有一宽峰,它是由O—H和N—H的伸缩振动产生[54]. 相比于纳米线,纳米线-H(图9b)两处Brønsted酸性位上的特征峰(1435 cm−1和1407 cm−1)强度有明显提升,而1541 cm−1处的Lewis酸位上的特征峰强度较低,已几乎不可见.值得注意的是,纳米线-H的表面在
1202 cm−1处出现了一个明显的新峰,且强度很高,它被指认为是配位连接在Lewis酸位点上的NH3[55 − 56]. 纳米线的Lewis酸性位和Brønsted酸性位吸收峰强度都相对较弱,说明纳米线对NH3的物理吸附和化学吸附能力都较差,此结果与纳米线NH3吸附容量只有0.15 mg·g−1相符. 而纳米线-H酸性位吸收峰更多且更强,表明其酸性位种类和数量更多,对NH3的吸附能力更强. 以上结果证实了表面酸性位的数量和强度对NH3吸附至关重要,这与NH3-TPD的分析结果一致. 另一方面,为进一步对比分析吸附为NH3的Lewis酸性位(1541 cm−1和1202 cm−1)和吸附为NH4+的Brønsted酸性位(1454 cm−1和1407 cm−1)的强度与变化,对不同时刻的原位红外吸收峰峰高及其增长率做了计算,结果如图9c、d所示. 可以发现,无论是在Lewis还是Brønsted酸位上,纳米线-H上的吸附物种数量都要显著高于纳米线上的吸附物种含量. 同样的,其NH3吸附物种形成速率,也是呈现类似的现象. 这表明,纳米线-H的表面酸性位点对NH3的吸附能力较强. 综上,原位红外的结果再次印证了更多更强的表面酸性位是纳米线-H优异氨气吸附性能的主要原因. -
1)通过水热法制备了四种不同形貌结构的α-MnO2(纳米线、纳米管、纳米棒和纳米花),并对其进行了表面酸性位强化. 其中酸性位强化后的α-MnO2纳米线-H在室温下表现出最佳的NH3吸附能力,吸附容量达到12.06 mg·g−1,是强化前(0.15 mg·g−1)的80倍. 在相同实验条件下,其吸附能力是未改性商业碳基材料(竹炭、椰壳活性炭和煤基活性炭)的6—12倍.
2)探究了吸附温度、NH3初始浓度和吸附剂用量对纳米线-H的NH3吸附性能的影响. 结果表明,NH3的吸附性能会随着吸附温度的上升而下降,而初始浓度和吸附剂用量对NH3吸附性能没有显著影响.
3)表征结果表明,酸性位强化后的纳米线-H上会产生更多的氧空位,其为潜在的Brønsted酸性位点. NH3-TPD和原位红外分析发现丰富的表面酸性位点是纳米线-H具有优异吸附性能的决定性原因,表面Lewis酸性位点和Brønsted酸性位点的增强是提高纳米线-H的NH3吸附性能的关键.
表面酸性位强化不同形貌α-MnO2对氨气的吸附性能研究
Study on the ammonia adsorption performance of α-MnO2 with different morphologies enhanced by surface acidic sites
-
摘要: 氨气(NH3)是常见的空气污染物之一,不仅导致各种环境问题还严重影响人体健康. 开发高性能氨气吸附材料,对于提高空气质量和保障人体健康具有重要意义. 本研究通过水热法制备了4种不同形貌结构的α-MnO2(纳米线、纳米管、纳米棒和纳米花),并进一步对其进行了表面酸性位强化,探究了其对NH3的吸附性能和吸附机制. 研究发现,酸性位强化的α-MnO2纳米线在室温下表现出最佳的NH3吸附能力,NH3吸附容量达到12.06 mg·g−1,是强化前(0.15 mg·g−1)的80倍,是相同条件下未改性商业碳基材料的6—12倍. 此外,还考察了吸附温度、初始NH3浓度、吸附剂用量对其吸附性能的影响. 最后,结合XRD、FESEM、BET、XPS、NH3-TPD和原位红外(in-situ DRIFTS)等对吸附剂进行了表征分析,结果表明丰富的表面酸性位点是酸性位强化α-MnO2纳米线具有优异吸附性能的决定性原因,表面Lewis酸性位点和Brønsted酸性位点的增强是提高NH3吸附性能的关键. 研究结果可为开发高效NH3吸附材料提供借鉴参考,为氨减排控制研究提供理论依据和技术支撑.Abstract: Ammonia (NH3) is one of the common air pollutants, which not only causes various environmental problems but also seriously threatens human health. Developing high-performance adsorbents for NH3 is of great significance for improving air quality and ensuring human health. In this study, α-MnO2 with four different morphologies (nanowire, nanotube, nanorod and nanoflower) were prepared by hydrothermal method, and their surface acidic sites were further strengthened to explore their adsorption performance and mechanism for NH3. It was found that the α-MnO2 nanowire strengthened by acidic sites exhibited the best NH3 adsorption capacity at room temperature, and the adsorption capacity reached 12.06 mg·g−1, which was 80 times higher than before strengthening (0.15 mg·g−1), and 6—12 times higher than unmodified commercial carbon-based materials under the same conditions. In addition, the effects of adsorption temperature, initial NH3 concentration and adsorbent dosage on the adsorption performance were also investigated. Finally, the adsorbents were characterized by XRD, FESEM, BET, XPS, NH3-TPD and in-situ DRIFTS. The results show that the abundant surface acidic sites are the decisive reason for the excellent adsorption performance of surface acidic sites strengthened α-MnO2. The enhancement of surface Lewis acidic sites and Brønsted acidic sites is the key to improve the NH3 adsorption performance. This research results can provide reference for the development of efficient NH3 adsorption materials, and provide theoretical basis and technical support for the research of emission reduction control of NH3.
-
Key words:
- ammonia adsorption /
- α-MnO2 /
- acidic site strengthening /
- adsorption mechanism /
- in-situ infrared.
-

-
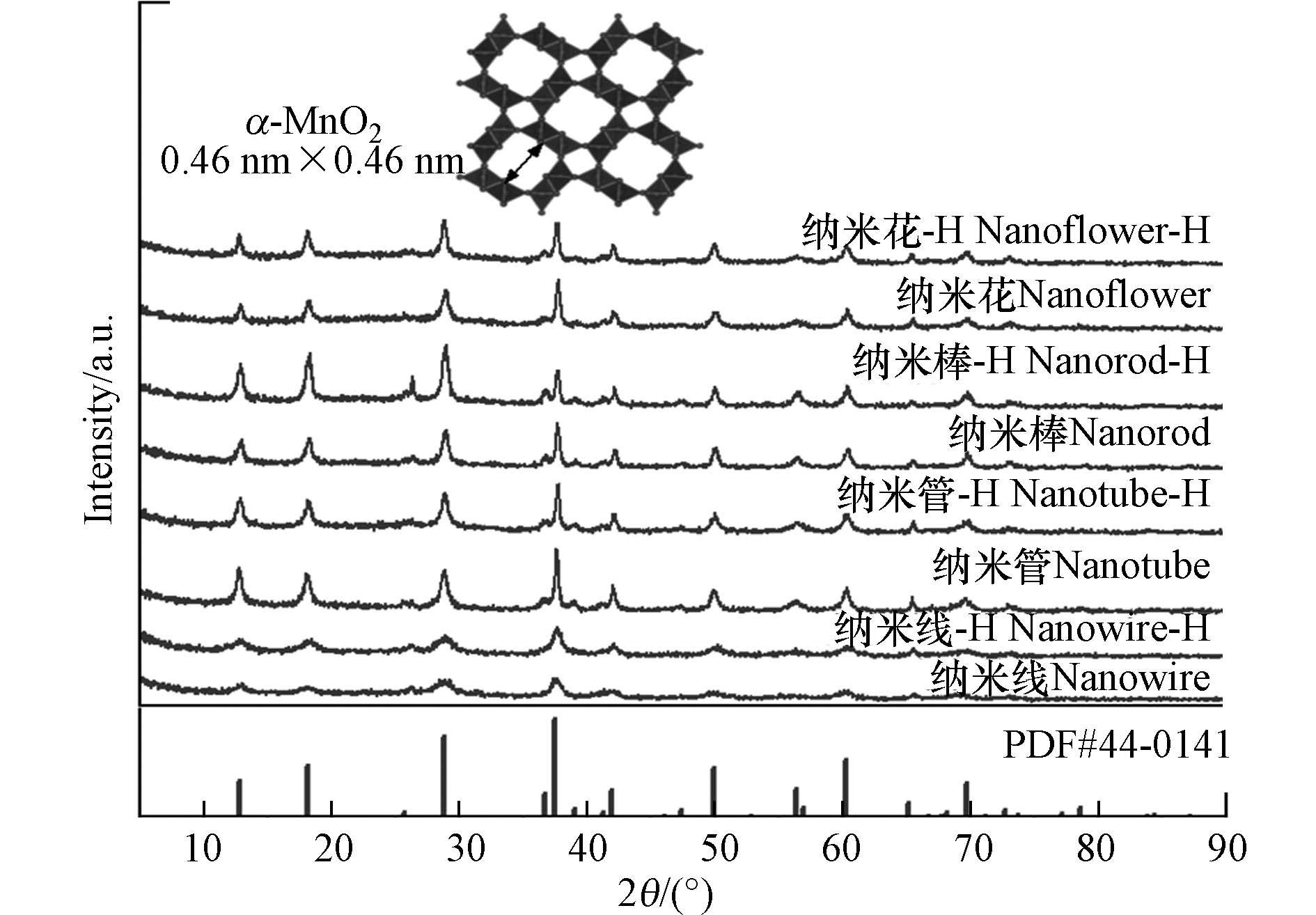
图 1 不同形貌α-MnO2的XRD谱图和晶体结构示意图
Figure 1. XRD patterns and crystal structure diagram of α-MnO2 with different morphologies

图 2 酸性位强化前后不同形貌α-MnO2的FESEM图
Figure 2. FESEM images of α-MnO2 with different morphologies before and after acidic site strengthening

图 3 不同形貌结构及酸性位强化α-MnO2的(a)NH3穿透曲线和(b)吸附容量
Figure 3. (a) Ammonia breakthrough curves and (b) adsorption capacities of different morphologies and acidic sites enhanced α-MnO2 (Initial NH3 concentration = 214 mg·m-3, ambient temperature = 25 ℃, adsorbent dosage = 100 mg)

图 4 (a, b)不同吸附温度(初始NH3浓度 = 209 mg·m−3,吸附剂用量=100 mg),(c, d)不同NH3浓度(环境温度=25 ℃,吸附剂用量=100 mg),(e, f)不同吸附剂用量(初始NH3浓度 = 209 mg·m−3,环境温度=25 ℃)下α-MnO2纳米线-H的NH3穿透曲线和吸附容量
Figure 4. Ammonia breakthrough curves and adsorption capacities of α-MnO2 nanowire-H under (a, b)different adsorption temperature (Initial NH3 concentration = 209 mg·m−3, adsorbent dosage = 100 mg), (c, d)different NH3 concentration (ambient temperature = 25°C, adsorbent dosage = 100 mg), (e, f)different adsorbent dosage (Initial NH3 concentration = 209 mg·m−3, ambient temperature = 25°C)
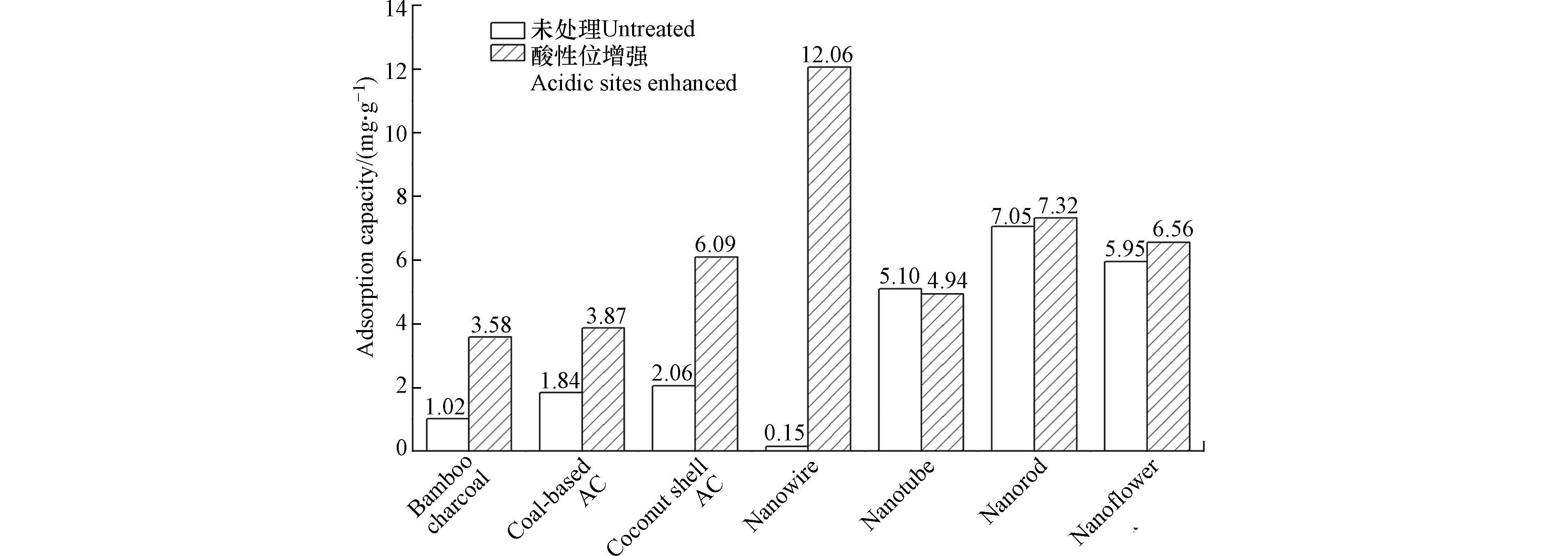
图 5 不同形貌结构的α-MnO2与三种常见的商业碳基材料对NH3的吸附容量对比
Figure 5. Comparison of NH3 adsorption capacities between α-MnO2 with different morphologies and three common commercial carbon-based materials
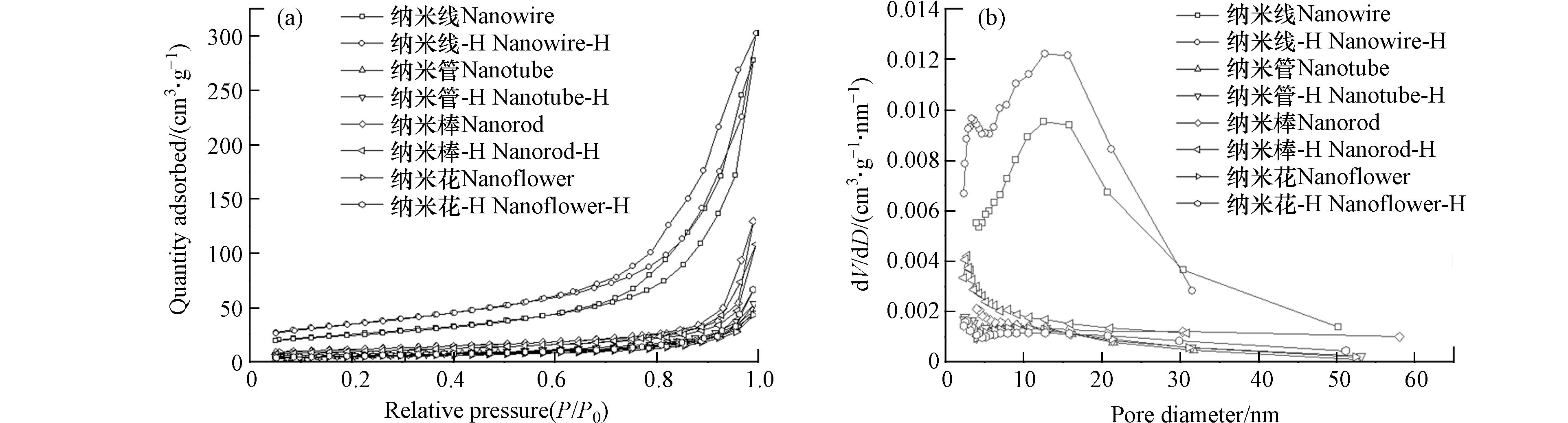
图 6 酸性位强化前后不同形貌α-MnO2的(a)N2吸附-脱附等温线和(b)孔径分布曲线
Figure 6. (a)N2 adsorption-desorption isotherms and (b)pore size distributions of α-MnO2 with different morphologies before and after acidic site strengthening

图 7 纳米线与纳米线-H的XPS结果 (a)Mn 3s, (b)O 1s, (c)各种氧物种含量以及表面吸附氧与晶格氧的比例, (d) K 2p
Figure 7. XPS results of nanowire and nanowire-H : (a) Mn 3s, (b) O 1s, (c) the content of various oxygen species and the ratio of surface adsorbed oxygen to lattice oxygen, (d) K 2p

图 9 (a, b)α-MnO2纳米线与纳米线-H暴露于NH3中不同时间的原位红外光谱图; (c, d)Lewis酸性位与Brønsted酸性位上NH3吸附物种的强度和形成速率随暴露时间的变化曲线
Figure 9. (a, b) In-situ DRIFTS of α-MnO2 nanowire and nanowire-H exposed to NH3 at different time; (c, d) The intensity and formation rate of NH3 adsorbed species on Lewis and Brønsted acidic sites as a function of exposure time
表 1 不同形貌结构α-MnO2的BET分析结果
Table 1. BET analysis of α-MnO2 with different morphologies
吸附剂
AbsorbentsSBET/(m2·g−1) 孔体积/(cm3·g−1)
Pore volume平均孔径/nm
Average pore size纳米线
nanowire88.21 0.27 10.04 纳米线-H
nanowire-H126.86 0.40 12.92 纳米管
nanotube24.17 0.05 7.78 纳米管-H
nanotube-H25.89 0.05 14.42 纳米棒
nanorod39.95 0.11 10.56 纳米棒-H
nanorod-H41.94 0.17 14.87 纳米花
nanoflower17.55 0.05 8.95 纳米花-H
nanoflower-H21.27 0.06 20.47  下载: 导出CSV
下载: 导出CSV
表 2 纳米线与纳米线-H的XPS分析结果
Table 2. XPS analysis results of nanowire and nanowire-H
吸附剂
AdsorbentsK/Mn Oads/Olatt AOS* 纳米线
nanowire0.81 0.30 3.65 纳米线-H
nanowire-H0.17 0.40 3.63 注:*根据XPS中Mn 3s的结合能差(ΔE),Mn的平均氧化态(AOS)可通过经验公式(AOS=8.956−1.126×ΔE)计算.
Note:* The average oxidation state (AOS) of Mn was calculated according to the binding energy difference (ΔE) of Mn 3s in XPS through an empirical formula (AOS=8.956−1.126×ΔE).
下载: 导出CSV
-
[1] 闫凤越, 李伟芳, 王亘, 等. 天津市工业区氨污染特征及感官特性评价[J]. 环境化学, 2019, 38(11): 2505-2509. doi: 10.7524/j.issn.0254-6108.2019031401 YAN F Y, LI W F, WANG G, et al. Study on pollution characteristics and sensory of NH3 in dustrial site of Tianjin City[J]. Environmental Chemistry, 2019, 38(11): 2505-2509 (in Chinese). doi: 10.7524/j.issn.0254-6108.2019031401
[2] 陈沛坤, 张袁斌, 崔希利, 等. 氨气深度脱除材料与技术研究进展[J]. 化工进展, 2021, 40(7): 3957-3975. CHEN P K, ZHANG Y B, CUI X L, et al. Progress in materials and technologies for deep removal of ammonia gas[J]. Chemical Industry and Engineering Progress, 2021, 40(7): 3957-3975 (in Chinese).
[3] CHEN S H, CHENG M M, GUO Z, et al. Enhanced atmospheric ammonia (NH3) pollution in China from 2008 to 2016: Evidence from a combination of observations and emissions[J]. Environmental Pollution, 2020, 263: 114421. doi: 10.1016/j.envpol.2020.114421 [4] FU X, WANG S X, XING J, et al. Increasing ammonia concentrations reduce the effectiveness of particle pollution control achieved via SO2 and NO x emissions reduction in East China[J]. Environmental Science & Technology Letters, 2017, 4(6): 221-227. [5] KONG L, TANG X, ZHU J, et al. Improved inversion of monthly ammonia emissions in China based on the Chinese ammonia monitoring network and ensemble Kalman filter[J]. Environmental Science & Technology, 2019, 53(21): 12529-12538. [6] CHANG Y H, ZOU Z, ZHANG Y L, et al. Assessing contributions of agricultural and nonagricultural emissions to atmospheric ammonia in a Chinese megacity[J]. Environmental Science & Technology, 2019, 53(4): 1822-1833. [7] 张小艺, 林伟立, 马志强, 等. 室内氨气浓度变化特征及其环境意义[J]. 环境化学, 2021, 40(10): 3270-3278. doi: 10.7524/j.issn.0254-6108.2020060501 ZHANG X Y, LIN W L, MA Z Q, et al. Variations in indoor ammonia concentration and its environmental significance[J]. Environmental Chemistry, 2021, 40(10): 3270-3278 (in Chinese). doi: 10.7524/j.issn.0254-6108.2020060501
[8] 谭静瑶, 王丽涛, 刘振通, 等. 邯郸市NH3污染特征及其在PM2.5形成中的作用[J]. 环境化学, 2021, 40(7): 2035-2046. doi: 10.7524/j.issn.0254-6108.2020120302 TAN J Y, WANG L T, LIU Z T, et al. NH3 pollution and its role in PM2.5 pollution in Handan, China[J]. Environmental Chemistry, 2021, 40(7): 2035-2046 (in Chinese). doi: 10.7524/j.issn.0254-6108.2020120302
[9] WU L B, REN H, WANG P, et al. Aerosol ammonium in the urban boundary layer in Beijing: Insights from nitrogen isotope ratios and simulations in summer 2015[J]. Environmental Science & Technology Letters, 2019, 6(7): 389-395. [10] HUANG R J, ZHANG Y L, BOZZETTI C, et al. High secondary aerosol contribution to particulate pollution during haze events in China[J]. Nature, 2014, 514: 218-222. doi: 10.1038/nature13774 [11] 张长斌. 室内空气污染物催化氧化研究[J]. 环境化学, 2015, 34(5): 817-823. doi: 10.7524/j.issn.0254-6108.2015.05.2015010509 ZHANG C B. Study of catalytic oxidation of indoor air pollutants[J]. Environmental Chemistry, 2015, 34(5): 817-823 (in Chinese). doi: 10.7524/j.issn.0254-6108.2015.05.2015010509
[12] BERHE GEBREEGZIABHER T, WANG S, NAM H. Adsorption of H2S, NH3 and TMA from indoor air using porous corncob activated carbon: Isotherm and kinetics study[J]. Journal of Environmental Chemical Engineering, 2019, 7(4): 103234. doi: 10.1016/j.jece.2019.103234 [13] VIKRANT K, KIM K H, DONG F, et al. Photocatalytic platforms for removal of ammonia from gaseous and aqueous matrixes: Status and challenges[J]. ACS Catalysis, 2020, 10(15): 8683-8716. doi: 10.1021/acscatal.0c02163 [14] DAVID A, PANCHARATNA K. Effects of acetaminophen (paracetamol) in the embryonic development of zebrafish, Danio rerio[J]. Journal of Applied Toxicology: JAT, 2009, 29(7): 597-602. doi: 10.1002/jat.1446 [15] KATZ M J, HOWARTH A J, MOGHADAM P Z, et al. High volumetric uptake of ammonia using Cu-MOF-74/Cu-CPO-27[J]. Dalton Transactions, 2016, 45(10): 4150-4153. doi: 10.1039/C5DT03436A [16] 赵晨阳, 李洪枚, 魏源送, 等. 翻堆频率对猪粪条垛堆肥过程温室气体和氨气排放的影响[J]. 环境科学, 2014, 35(2): 533-540. ZHAO C Y, LI H M, WEI Y S, et al. Effects of turning frequency on emission of greenhouse gas and ammonia during swine manure windrow composting[J]. Environmental Science, 2014, 35(2): 533-540 (in Chinese).
[17] BEHERA S N, SHARMA M, ANEJA V P, et al. Ammonia in the atmosphere: A review on emission sources, atmospheric chemistry and deposition on terrestrial bodies[J]. Environmental Science and Pollution Research, 2013, 20(11): 8092-8131. doi: 10.1007/s11356-013-2051-9 [18] SAARELA K, JÄRNSTRÖM H. Indoor air quality in new residential buildings and behaviour of materials in structures[J]. Indoor and Built Environment, 2003, 12(4): 243-247. doi: 10.1177/1420326X03035096 [19] LI M Z, WESCHLER C J, BEKÖ G, et al. Human ammonia emission rates under various indoor environmental conditions[J]. Environmental Science & Technology, 2020, 54(9): 5419-5428. [20] GAO F Y, LIU Y Y, SANI Z, et al. Advances in selective catalytic oxidation of ammonia (NH3-SCO) to dinitrogen in excess oxygen: A review on typical catalysts, catalytic performances and reaction mechanisms[J]. Journal of Environmental Chemical Engineering, 2021, 9(1): 104575. doi: 10.1016/j.jece.2020.104575 [21] CHMIELARZ L, JABŁOŃSKA M. Advances in selective catalytic oxidation of ammonia to dinitrogen: A review[J]. RSC Advances, 2015, 5(54): 43408-43431. doi: 10.1039/C5RA03218K [22] HAN B, BUTTERLY C, ZHANG W, et al. Adsorbent materials for ammonium and ammonia removal: A review[J]. Journal of Cleaner Production, 2021, 283: 124611. doi: 10.1016/j.jclepro.2020.124611 [23] HUANG C C, LI H S, CHEN C H. Effect of surface acidic oxides of activated carbon on adsorption of ammonia[J]. Journal of Hazardous Materials, 2008, 159(2/3): 523-527. [24] GONÇALVES M, SÁNCHEZ-GARCÍA L, de OLIVEIRA JARDIM E, et al. Ammonia removal using activated carbons: Effect of the surface chemistry in dry and moist conditions[J]. Environmental Science & Technology, 2011, 45(24): 10605-10610. [25] CANALS-BATLLE C, ROS A, LILLO-RÓDENAS M A, et al. Carbonaceous adsorbents for NH3 removal at room temperature[J]. Carbon, 2008, 46(1): 176-178. doi: 10.1016/j.carbon.2007.10.017 [26] TAKAHASHI A, MINAMI K, NODA K, et al. Trace ammonia removal from air by selective adsorbents reusable with water[J]. ACS Applied Materials & Interfaces, 2020, 12(13): 15115-15119. [27] REZAEI E, SCHLAGETER B, NEMATI M, et al. Evaluation of metal oxide nanoparticles for adsorption of gas phase ammonia[J]. Journal of Environmental Chemical Engineering, 2017, 5(1): 422-431. doi: 10.1016/j.jece.2016.12.026 [28] REZAEI E, AZAR R, NEMATI M, et al. Gas phase adsorption of ammonia using nano TiO2-activated carbon composites–Effect of TiO2 loading and composite characterization[J]. Journal of Environmental Chemical Engineering, 2017, 5(6): 5902-5911. doi: 10.1016/j.jece.2017.11.010 [29] ZHENG W H, HU J T, RAPPEPORT S, et al. Activated carbon fiber composites for gas phase ammonia adsorption[J]. Microporous and Mesoporous Materials, 2016, 234: 146-154. doi: 10.1016/j.micromeso.2016.07.011 [30] ZHANG D L, SHEN Y J, DING J T, et al. A combined experimental and computational study on the adsorption sites of zinc-based MOFs for efficient ammonia capture[J]. Molecules, 2022, 27(17): 5615. doi: 10.3390/molecules27175615 [31] GRANT GLOVER T, PETERSON G W, SCHINDLER B J, et al. MOF-74 building unit has a direct impact on toxic gas adsorption[J]. Chemical Engineering Science, 2011, 66(2): 163-170. doi: 10.1016/j.ces.2010.10.002 [32] DEHMANI Y, DRIDI D, LAMHASNI T, et al. Review of phenol adsorption on transition metal oxides and other adsorbents[J]. Journal of Water Process Engineering, 2022, 49: 102965. doi: 10.1016/j.jwpe.2022.102965 [33] 张传巧, 陈静, 吴秋月, 等. Ce-Mn复合氧化物对As(V)的吸附行为与机制[J]. 环境化学, 2020, 39(12): 3542-3551. doi: 10.7524/j.issn.0254-6108.2019090605 ZHANG C Q, CHEN J, WU Q Y, et al. Adsorption of As (V) on Ce-Mn binary oxide: Behavior and mechanism[J]. Environmental Chemistry, 2020, 39(12): 3542-3551 (in Chinese). doi: 10.7524/j.issn.0254-6108.2019090605
[34] 陈晨, 李北罡. Fe3O4@SA/Ce微球的表征及对染料的吸附特性[J]. 环境化学, 2021, 40(3): 799-807. doi: 10.7524/j.issn.0254-6108.2019102601 CHEN C, LI B G. Characterization of Fe3O4@SA/Ce microspheres and their adsorption characteristics for direct dyes[J]. Environmental Chemistry, 2021, 40(3): 799-807 (in Chinese). doi: 10.7524/j.issn.0254-6108.2019102601
[35] ZHOU Y, WU Z, DING D N, et al. Tunnel structured manganese dioxides for the gaseous ammonia adsorption and its regeneration performance[J]. Separation and Purification Technology, 2022, 284: 120252. doi: 10.1016/j.seppur.2021.120252 [36] RONG S P, ZHANG P Y, LIU F, et al. Engineering crystal facet of α-MnO2 nanowire for highly efficient catalytic oxidation of carcinogenic airborne formaldehyde[J]. ACS Catalysis, 2018, 8(4): 3435-3446. doi: 10.1021/acscatal.8b00456 [37] XIAO W, XIA H, FUH J Y H, et al. Growth of single-crystal α-MnO2 nanotubes prepared by a hydrothermal route and their electrochemical properties[J]. Journal of Power Sources, 2009, 193(2): 935-938. doi: 10.1016/j.jpowsour.2009.03.073 [38] HE T H, SHAO D D, ZENG X S, et al. Harvesting the vibration energy of α-MnO2 nanostructures for complete catalytic oxidation of carcinogenic airborne formaldehyde at ambient temperature[J]. Chemosphere, 2020, 261: 127778. doi: 10.1016/j.chemosphere.2020.127778 [39] TOMPSETT D A, PARKER S C, ISLAM M S. Surface properties of α-MnO2: Relevance to catalytic and supercapacitor behaviour[J]. Journal of Materials Chemistry A, 2014, 2(37): 15509-15518. doi: 10.1039/C4TA00952E [40] HELMINEN J, HELENIUS J, PAATERO E, et al. Comparison of sorbents and isotherm models for NH3-gas separation by adsorption[J]. AIChE Journal, 2000, 46(8): 1541-1555. doi: 10.1002/aic.690460807 [41] 王海林. 改性柚皮基生物炭对氨气的吸附性能及其机理研究[D].重庆: 重庆大学, 2021. WANG H L. Adsorption performance of modified pomelo peel-based biochar for ammonia and its mechanism research[D]. Chongqing:Chongqing University, 2021 (in Chinese).
[42] HE W J, LU J L, ZHANG N, et al. Surface acidic sites strengthened core-shell HC@MnO2 for enhanced gaseous ammonia adsorption[J]. Chemosphere, 2023, 338: 139507. doi: 10.1016/j.chemosphere.2023.139507 [43] JIA J B, ZHANG P Y, CHEN L. Catalytic decomposition of gaseous ozone over manganese dioxides with different crystal structures[J]. Applied Catalysis B: Environmental, 2016, 189: 210-218. doi: 10.1016/j.apcatb.2016.02.055 [44] LIU Y, YANG W J, ZHANG P Y, et al. Nitric acid-treated birnessite-type MnO2: An efficient and hydrophobic material for humid ozone decomposition[J]. Applied Surface Science, 2018, 442: 640-649. doi: 10.1016/j.apsusc.2018.02.204 [45] ZHOU Y, RONG S P, XIE H F, et al. Enhancement of acidic sites in layered MnO2 for the highly efficient selective catalytic oxidation of gaseous ammonia[J]. Journal of Environmental Chemical Engineering, 2023, 11(2): 109480. doi: 10.1016/j.jece.2023.109480 [46] WANG F, DAI H X, DENG J G, et al. Manganese oxides with rod-, wire-, tube-, and flower-like morphologies: Highly effective catalysts for the removal of toluene[J]. Environmental Science & Technology, 2012, 46(7): 4034-4041. [47] YIN X L, HAN H M, GUNJI I, et al. NH3 adsorption on the Brönsted and Lewis acid sites of V2O5(010): A periodic density functional study[J]. The Journal of Physical Chemistry B, 1999, 103(22): 4701-4706. doi: 10.1021/jp990363p [48] ZHANG N Q, LI L C, GUO Y Z, et al. A MnO2-based catalyst with H2O resistance for NH3-SCR: Study of catalytic activity and reactants-H2O competitive adsorption[J]. Applied Catalysis B: Environmental, 2020, 270: 118860. doi: 10.1016/j.apcatb.2020.118860 [49] MARTINS G V A, BERLIER G, BISIO C, et al. Quantification of Brønsted acid sites in microporous catalysts by a combined FTIR and NH3-TPD study[J]. The Journal of Physical Chemistry C, 2008, 112(18): 7193-7200. doi: 10.1021/jp710613q [50] RONG S P, HE T H, ZHANG P Y. Self-assembly of MnO2 nanostructures into high purity three-dimensional framework for high efficiency formaldehyde mineralization[J]. Applied Catalysis B: Environmental, 2020, 267: 118375. doi: 10.1016/j.apcatb.2019.118375 [51] RONG S P, ZHANG P Y, YANG Y J, et al. MnO2 framework for instantaneous mineralization of carcinogenic airborne formaldehyde at room temperature[J]. ACS Catalysis, 2017, 7(2): 1057-1067. doi: 10.1021/acscatal.6b02833 [52] HUANG Z G, ZHU Z P, LIU Z Y, et al. Formation and reaction of ammonium sulfate salts on V2O5/AC catalyst during selective catalytic reduction of nitric oxide by ammonia at low temperatures[J]. Journal of Catalysis, 2003, 214(2): 213-219. doi: 10.1016/S0021-9517(02)00157-4 [53] XIE L L, GAO Q M, WU C D, et al. Rapid hydrothermal synthesis of bimetal cobalt nickel phosphate molecular sieve CoVSB-1 and its ammonia gas adsorption property[J]. Microporous and Mesoporous Materials, 2005, 86(1/2/3): 323-328. [54] PETIT C, MENDOZA B, BANDOSZ T J. Reactive adsorption of ammonia on Cu-based MOF/graphene composites[J]. Langmuir: the ACS Journal of Surfaces and Colloids, 2010, 26(19): 15302-15309. doi: 10.1021/la1021092 [55] SI Z C, WENG D, WU X D, et al. Synergistic effects between copper and tungsten on the structural and acidic properties of CuO x/WO x–ZrO2 catalyst[J]. Catalysis Science & Technology, 2011, 1(3): 453-461. [56] WANG F, HE G Z, ZHANG B, et al. Insights into the activation effect of H2 pretreatment on Ag/Al2O3 catalyst for the selective oxidation of ammonia[J]. ACS Catalysis, 2019, 9(2): 1437-1445. doi: 10.1021/acscatal.8b03744 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1267
- HTML全文浏览数: 1267
- PDF下载数: 40
- 施引文献: 0
