

 下载:
下载:

-
非均相芬顿反应体系中如何实现高pH范围内活化H2O2生成羟基自由基(·OH)一直是芬顿催化反应研究的重点,较高的pH导致水中H2O2稳定性降低、催化剂Fe2+沉淀以及·OH产生量减少且寿命变短[1]. 氧基氯化铁(FeOCl)作为一种在较宽pH范围具有高催化活性的Fe基非均相催化剂,其具有制备方法简单、表面催化机制独特以及可以重复利用的特点[2 − 4],大量研究证明FeOCl在中性环境下依然可以产生远多于传统FeSO4以及Fe2O3等催化剂的·OH[5],且对抗生素、双酚A、罗丹明B等多种污染物的处理中体现出较高的催化活性[6 − 9]. 然而在实际废水处理中发现很难达到实验室研究所取得的效果,FeOCl在实际废水体系中的催化效果以及对水中TOC的彻底矿化效果却少有关注,其中,实际废水pH缓冲性作为一个重要的干扰因素值得详细探讨.
制革废水经生化处理后的尾水中仍含有大量Na+、Ca2+、Cl−、SO42-、CO32-等离子盐及多酚、类腐殖质等有机污染物[10 − 13],由此造成废水具有较强的pH缓冲性,在传统芬顿进行深度处理中需消耗大量的酸用于降低系统的pH[14],FeOCl芬顿催化反应在高pH缓冲性的制革废水处理中进行实验研究对于其工程应用具有重要意义. 没食子酸(GA)作为单宁类鞣剂的水解产物,其酚羟基供电子能力较强,使其对变价金属Fe具有一定的还原作用[15 − 16],将其作为模拟物既可以了解其促进FeOCl芬顿催化反应效率的机制,也可与实验制革废水TOC矿化进行对比分析. 据此,本文以FeOCl为非均相芬顿催化剂,以制革生化尾水以及没食子酸模拟废水为对象,探究简单与复杂水质中不同的pH缓冲性对FeOCl非均相体系对TOC矿化效果的影响,通过监测芬顿反应过程中体系pH变化以及FeOCl的水解调节pH能力、芬顿反应过程中产生H+能力,获得适合制革废水FeOCl催化的pH调控机制,在此基础上探究影响FeOCl循环利用的因素及活化再生方法,以期为FeOCl在制革废水芬顿处理过程中减泥降成本提供一定的理论和实践依据.
-
供试废水为河北某制革企业生化处理后尾水(pH=7.5—8,TOC = 70—80 mg·L−1,COD=172—180 mg·L−1);模拟废水由没食子酸(GA)配制成80 mg·L−1的溶液,其TOC为 40—45 mg·L−1;过氧化氢(30%),六水合三氯化铁、七水合硫酸亚铁均为分析纯.
-
根据已有的FeOCl制备相关文献[17],采用较为稳定的部分热解法制备FeOCl,具体方法为:取适量FeCl3·6H2O置于玛瑙研钵中充分研磨10 min,将其盛于坩埚内并加盖,设定250 ℃为最佳煅烧温度,因文献报道中对升温速率设置不统一,本实验于马弗炉中分别以1 ℃·min−1、5 ℃·min−1、10 ℃·min−1的升温速率升温至250 ℃煅烧1 h. 自然冷却后用无水乙醇离心清洗3次以上去除多余的FeCl3,最后置于真空干燥箱中80 ℃干燥12 h得到红褐色粉末即为FeOCl,研磨充分后密封干燥保存备用.
-
实验通过控制初始pH、FeOCl投加量、H2O2投加量等参数对没食子酸溶液中TOC进行矿化进而确定最佳反应条件. 具体方法为:于100 mL模拟废水中,加入不同量的FeOCl粉末和H2O2,使用NaOH和H2SO4调节pH后,加入30% H2O2溶液启动芬顿反应,于不同时间取水样,经0.45 μm滤膜分离出固体后测定液体TOC浓度.
-
首先对模拟废水与实际废水进行了pH缓冲性测定,即分别取100 mL模拟废水或实际废水,测定其初始pH值,随后使用0.1 mol·L−1H2SO4溶液对两体系进行滴定,测定pH缓冲性. 然后分别用FeOCl粉末和FeSO4·7H2O,逐步加入不同剂量,均匀反应后测定体系pH随物质投加量的变化趋势.
取100 mL模拟废水或实际废水,使用NaOH和H2SO4调节pH,投加一定量的FeOCl或FeSO4·7H2O搅拌均匀,加入30%H2O2溶液启动催化体系,于不同时间取样,经0.45 μm滤膜分离后测定TOC浓度以及反应过程中的pH值变化.
-
于100 mL初始pH为4的生化尾水中投加1 g FeOCl催化剂并加入H2O2启动反应,每次反应结束后进行离心,将分离出的催化剂重新投入废水中进行催化反应,重复5次. 再生实验则是在离心后,将催化剂置于100 mL 0.05 mol·L−1的HCl中超声5 min,随后离心分离出催化剂重新投入废水中进行催化反应,重复10次.
-
上述实验中,FeOCl晶体结构与形貌分析使用日本Rigaku公司SmartLab9KW型X射线衍射仪(XRD)、德国ZEISS Sigma 300扫描电子显微镜(SEM)进行表征. 催化反应前后TOC变化:使用德国Elementar公司Liqui TOC Ⅱ型测定仪测定. 催化反应活性氧自由基使用德国Bruker EMXplus-6/1顺磁共振波谱仪检测. 体系反应过程中pH值由德国Sartorious PB-10型pH计测定记录.
-
图1为不同升温速率条件下制备获得的FeOCl的SEM图像. 如图1所示,1 ℃·min−1升温速率条件下,FeOCl呈现不规则条形薄片状结构,片层结构未完全形成;升温速率提升至5 ℃·min−1时的FeOCl呈现出明显的片层状堆叠结构,表面逐渐光滑;进一步提升升温速率至10 ℃·min−1,FeOCl的纳米片结构更加清晰,总体结构由表面光滑的纳米薄片堆叠而成,且薄片厚度较为均匀,平均厚度在80—100 nm.
由不同升温速率下FeOCl晶体X射线衍射谱图可以看出(图2a),所有的FeOCl都在10.8°、26.0°、35.5°和38.9°产生了明显的峰,这与纯相FeOCl的标准卡片(PDF#72-0619)一致,表明FeOCl通过部分热解法成功制备. 同时发现,随着升温速率增加,峰强明显降低,说明FeOCl结晶度出现下降. 各谱图中伴有少量的低矮杂峰,可能是局部受热不均产生的少量α-Fe2O3[18].
将3种FeOCl样品在初始pH4、FeOCl投加量800 mg·L−1、H2O2投加量6 mmol·L−1条件下对80 mg·L−1的没食子酸溶液中TOC去除效果进行分析(图2b),可以看出,在反应完成后,1 ℃·min−1、5 ℃·min−1和10 ℃·min−1升温速率下制备的FeOCl的TOC矿化率分别为55.32%、63.01%和68.34%. 说明制备FeOCl时快速升温有助于提高FeOCl催化性能. 综合图1结果,快速升温有利于催化剂纳米片状结构的形成,使其具有较大的比表面积,可将更多活性Fe位点暴露在体系中与污染物和H2O2接触,从而更高效地产生·OH以矿化污染物.
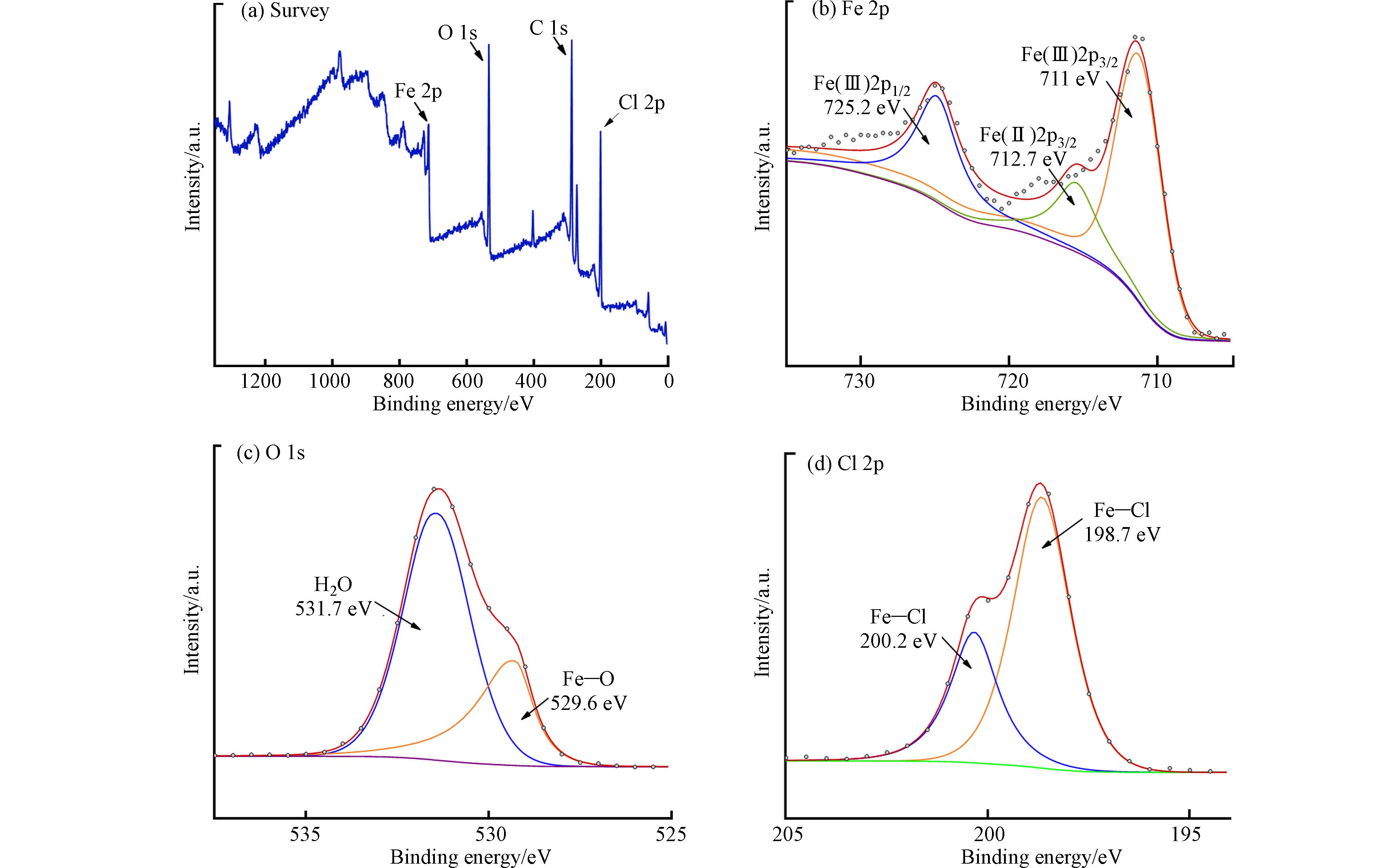
通过XPS光谱对催化剂元素组成及各元素配位环境进行表征,图3可以看出,Fe、O、Cl等元素均存在于催化剂当中;在结合能725 eV与711 eV处分别产生对应于Fe(Ⅲ)中2p3/2和2p1/2的峰,表明催化剂中Fe主要以三价的形态存在,在529 eV处产生的峰对应为Fe—O键,结合能198.7 eV和200.2 eV处对应为Fe—Cl键.
-
于pH4条件下设置了0.4、0.8、1.6、2、3、3.5、4、4.5 g·L−1等8个催化剂投加梯度以及1、3、6、9、12 mmol·L−1等5个H2O2浓度梯度进行正交实验,结果如图4所示. 当催化剂从0.4 g·L−1增加到3.5 g·L−1,H2O2投加浓度提升到6 mmol·L−1,GA溶液中TOC去除率最高达到了75.63%,继续投加催化剂和H2O2后TOC去除率没有明显提升. 表明H2O2在实验浓度下已达到临界值,H2O2与材料的接触几率以及·OH产率不再受影响. 过高H2O2浓度可导致其无效分解且与·OH自由基反应生成活性较低的·O2H自由基[19 − 20],一定程度地降低体系的催化效率. 本文后续实验选择6 mmol·L−1为H2O2最适投加浓度、选取3.5 g·L−1为FeOCl最佳投加量,即图b响应曲面中A点所代表的投加量.
-
实验采用电子顺磁共振(EPR)对不同GA浓度下·OH强度进行了检测,测试过程使用DMPO(5,5-二甲基-1-吡咯啉-N-氧化物)作为捕获剂对·OH进行捕获,结果如图5a所示. 从图5a中可以看出各催化体系中由DMPO-·OH产生的1:2:2:1独特特征峰,由此判断FeOCl体系产生了·OH自由基. 同时发现,在FeOCl/H2O2芬顿体系中添加了少量GA后,·OH产生量明显增高,提升GA浓度后,反应所消耗的H2O2量显著增加,·OH产量进一步提升,这与图5b中布鲁克绝对定量·OH浓度变化规律相吻合. 这一结果表明,GA可以有效促进FeOCl体系中H2O2向·OH的转化. 由图5c和图5d可看出,反应结束后,浓度为80 mg·L−1的GA体系中FeOCl表面Fe2+占比相较于浓度为20 mg·L−1的GA体系显著增加,而Fe3+显著减少.
结合相关文献[15 − 16],其原因为GA具有较强的还原性,在与FeOCl表面以及溶液中的Fe3+接触后,可以通过与铁离子配位的形式进行GA与Fe3+之间的电子传递,使得Fe3+向Fe2+转化,从而促进H2O2向·OH的高效转化. 综上所述,GA这类还原性物质主要通过促进FeOCl表面Fe3+向Fe2+转化,使得单位时间内更充分地消耗H2O2从而产生更多的·OH,进而实现芬顿体的矿化效率的显著提升.
-
由于FeOCl具有水解产生H+的能力,使得废水的pH在反应过程中不断降低. 同时,在H2O2加入后,FeOCl表面Fe3+会吸附H2O2发生脱氢反应释放出H+[3, 21],并且随着芬顿反应的进行,有机物会被氧化分解并产生少量的小分子有机酸[10, 22],这也使得整个体系的pH逐渐向利于芬顿反应进行的酸性环境转变. 为确定废水缓冲性以及方便对FeOCl引入的H+进行计算,分别使用0.1 mol·L−1的H2SO4溶液、FeOCl粉末以及FeSO4·7H2O颗粒对GA模拟废水以及实际生化尾水的缓冲能力进行测定,结果如图6所示. 由图6可知,模拟废水与实际生化尾水的酸缓冲性具有显著差异,模拟废水体系简单,溶质单一,使得pH随着酸性物质的引入会迅速降低. 实际废水成分复杂,对于酸性物质具有较高的缓冲能力,使得其pH变化缓慢,相较于模拟废水需要投加3—4倍甚至更多量的酸性物质才能到相同pH条件.
-
分别设置两组GA模拟体系,第1组为反应前将3.5 g·L−1 FeOCl加入到GA溶液后调整到不同初始pH值,以排除FeOCl自身水解产生的H+,第2组为将GA溶液调节好pH后再向体系中投加3.5 g·L−1 FeOCl. 如图6a所示,投加3.5 g·L−1的 FeOCl可通过水解使100 mL GA模拟废水pH由7.0下降到3.75,结合图6a中H2SO4滴定曲线,计算得FeOCl水解可产生5×10−4 mol·L−1的H+. 由图7a核算得到,3.5 g·L−1 FeOCl条件下芬顿反应过程中产生了2.6×10−4 mol·L−1的H+. 两者合计共产生7.6×10−4 mol·L−1的H+,此结果与图7b中pH值降解所需要的H+浓度相吻合. FeOCl通过水解与芬顿过程引入H+的数量比约为2:1. 同时由图7b可以看出,GA溶液即使在初始pH7条件下FeOCl催化依然可使TOC去除率达到70%左右. 即对于单一GA体系,利用FeOCl自身的水解以及芬顿反应产生的H+可实现中性环境下的高效芬顿催化.
-
在模拟废水中验证了通过FeOCl自身调节pH的可行性,因此期望利用这一特性对废水pH进行调节,以减少在废水处理前人为添加酸的量,有效降低废水处理成本. 由于实际废水中TOC含量在75—85 mg·L−1左右,约为没食子酸模拟废水的1.5—2倍,且实际废水成分更为复杂,处理难度更大,因此在后续对实际废水的处理实验中,将FeOCl投加量提升至10 g·L−1,H2O2投加量提升至18 mmol·L−1,以便取得更好的处理效果并将其效果与常规芬顿处理效果进行对比.
由图8a、b对比看出,在初始pH=4、5、6和7时,TOC矿化率分别为50.8%、48.75%、32.09%以及18.24 %,相比于同pH条件下的传统芬顿体系分别提高21.09 %、21.93 %、9.33 %和2.65 %,在初始pH=6时即可达到传统芬顿在pH=4的最适条件下所能达到的矿化效果,这表明FeOCl相较于传统芬顿具有较宽的pH适应范围以及更显著的TOC矿化效果. 这是因为在同等体系环境条件下,FeOCl能够在单位时间内产生几倍于其它催化剂的·OH,使其催化效率更高[23].
图8c、d中显示,pH≤5的体系pH迅速下降后则保持在3—3.5,而在初始pH为6和7时,pH在经过前5 min的迅速降低后缓慢回升至初始pH值,由于前5 min内pH的迅速降低,体系环境总体有利于芬顿反应进行,因此TOC在前5 min迅速发生矿化,后随着pH回升而减缓,这表明实际废水的缓冲性对芬顿体系具有较大的影响. 结合图6b可知,将实际废水从pH7调节到pH4需要通过添加0.1 mol·L−1 H2SO4引入约6×10−3 mol·L−1的H+,而10 g·L−1 FeOCl通过水解只能将实际废水从pH7调节到约pH6.4,利用H2SO4引入量换算得到H+引入量仅有2.5×10−3 mol·L−1,即使加上按照2.3.2节中比例计算得出芬顿反应引入的1.25×10−3 mol·L−1 H+,共计3.75×10−3 mol·L−1 H+,也仍有2.25×10−3 mol·L−1的H+缺口,由于FeOCl自身产酸性能难以克服pH6和pH7时实际废水较高的缓冲性,使得pH在前5 min迅速下降后重新调节回升至初始的稳定pH状态.
虽然图8a数据以及相关文献表明,FeOCl可以在中性以及弱酸性环境进行TOC矿化,且矿化效果远高于同条件下传统芬顿,但综合相关研究所得结论,pH≤5才是发挥FeOCl芬顿体系最佳性能的环境. 首先,在弱酸和中性环境中并不利于FeOCl体系中发挥主要氧化作用的·OH[24]的生成. Sun等通过实验测得中性理想条件下,0.2 g·L−1 FeOCl 与15 mmol·L−1 H2O2在反应开始1 min内的·OH累积量约为11 μmol·L−1,但中性相较于酸性环境,·OH数量显著降低且·OH寿命较短,累积量仅为酸性条件的1/3,因此酸性条件依旧是最适合芬顿反应进行的环境[23]. 其次,pH升高会促使FeOCl表面铁配合物的生成,占用了过多的表面铁活性位点,使得催化活性降低[25]. 最后,过高的体系pH会影响H2O2的稳定性,在较高的pH环境下会导致H2O2自我分解,H2O2有效浓度的降低导致芬顿反应效率整体降低[26]. 这些都导致芬顿催化体系效率降低从而难以实现有机物的迅速矿化. 因此,从发挥FeOCl芬顿整体最大性能的角度出发,体系pH应保持在5以下,由于FeOCl自身水解以及芬顿过程中产H+的特性,可有效减少调酸过程中酸的消耗.
-
循环催化能力是评价非均相催化剂性能的主要因素,通过将FeOCl在实际废水中直接循环催化5次来探究其循环性能,结果如图9a所示:FeOCl在直接循环3次时对TOC的矿化率基本与传统芬顿持平,第4次与第5次时矿化率远低于传统芬顿,这说明FeOCl在直接回用中催化效率会逐渐降低. 结合XPS、SEM以及红外光谱等对其活性降低原因进行分析,循环5次后FeOCl的XPS如图9b所示,对比图3b所反映的反应前材料表面Fe元素价态,发现Fe2+峰面积显著减小,而依据FeOCl催化机理[3]以及已有研究显示[18],催化反应发生后FeOCl表面Fe2+含量应该相对增加,这说明FeOCl表面Fe3+在多次循环催化过程中向Fe2+转化的过程受阻. 通过SEM观察5次循环后材料表面,发现表面不再平滑,出现大量缺陷并且黏附有大量微小颗粒,推测为废水中有机物吸附于催化剂表面致使表面Fe活性位点减少. 反应前后材料红外光谱如图9d显示,反应后500 cm−1处Fe—O键产生的峰有所降低,说明过程中Fe元素有所流失,反应后
1079 cm−1产生较小的—COO—的伸缩振动峰,这可能为表面Fe3+与废水中有机物的羧基结合形成络合物[27],其余峰位并没有发生明显变化,推测过程中主要以物理吸附为主,同时发生羧基与Fe3+结合的化学吸附,随着循环次数的增加,表面附着的有机物越来越多,从而导致表面活性位点减少,致使FeOCl活性降低. 同时结合前文中提到的FeOCl自身的酸调节能力,推测在多次反应后,由于表面暴露的Fe活性位点减少以及Fe的流失,致使材料水解以及芬顿过程H+产量降低,使其抗缓冲能力减弱,一定程度限制其反应效率.为提高FeOCl的循环催化次数,使用酸洗协同超声的方式对FeOCl进行活化,结果如图10所示,通过这种方式可将FeOCl的循环次数提升至10次以上,有效提高了FeOCl的循环催化性能. 其原理为通过超声将物理吸附于材料表面的有机物脱离下来,0.05 mol·L−1盐酸则可以脱附化学结合在材料表面有机物. 同时超声以及盐酸对材料表面起到剥离与侵蚀作用,使得平滑表面变得凹凸不平,如图10(b)所示,这可以使材料暴露出更多活性位点从而有效提升材料后续的催化效果.
-
(1)本研究基于部分热解法制备了FeOCl催化剂,以250 ℃为最佳煅烧温度保持1 h,速率升温为10 ℃·min−1时,所得材料片层状结构生成稳定且分布均匀.
(2)FeOCl在宽泛的pH范围内展现出优异的TOC矿化能力. 对初始pH=4、5、6和7的实际废水,TOC矿化率分别为50.8%、48.75%、32.09%以及18.24%,均优于传统芬顿,在初始pH6时即可达到传统芬顿在pH4的最适条件下所能达到的矿化效果.
(3)FeOCl可通过自身水解以及芬顿反应过程产生H+对废水pH值进行调节. 最佳芬顿反应条件下,对GA溶液,FeOCl催化剂通过自身水解与芬顿反应生成约7.6×10−4 mol·L−1的H+,将体系pH调至3—4,对实际废水亦可引入约3.75×10−3 mol·L−1的H+,显著减少了外加酸的使用需求,有效降低了废水处理成本.
(4)FeOCl对有机物的物理吸附以及Fe3+与羧基的络合导致活性Fe位点减少,是催化剂逐渐失活的主要原因. 经超声与酸洗协同再生可使循环次数从3次提升至10次以上,其催化性能仍显著高于传统芬顿体系,展示出该催化剂在实际工程应用中优异的处理效果和长期稳定性.
FeOCl芬顿反应过程中废水pH缓冲性与TOC降解特征
Effect of pH buffering on TOC mineralization in FeOCl Fenton system
-
摘要: 摘 要FeOCl作为一种非均相芬顿催化剂,在宽泛的pH范围内表现出优异的催化能力,但其pH适应性受水质影响显著. 本研究采用部分热解法制备了FeOCl催化剂,并以制革生化尾水为对象,探讨了FeOCl芬顿反应过程中废水pH缓冲性对总有机碳(TOC)矿化效果的影响,并与没食子酸(GA)单一模拟物条件下的行为进行了比较. 研究表明,在最佳反应条件下(6 mmol·L−1 H2O2,3.5 g·L−1 FeOCl),FeOCl通过水解和芬顿过程可产生约7.6×10−4 mol·L−1的H+,使初始pH由中性快速降低至pH3—4之间,此时TOC矿化率可达70%左右. 对于实际废水,本工艺运行条件下,FeOCl可引入约3.75×10−3 mol·L−1的H+,仅需加酸补充2.25×10−3 mol·L−1的H+即可将pH调至4,在初始pH6时即可达到传统芬顿最适pH条件下的矿化效果. FeOCl在废水中对有机物的物理吸附以及Fe3+与羧基的络合导致活性Fe位点减少,是催化剂逐渐失活的主要原因. 通过酸洗协同超声处理,可以有效脱附有机物并暴露更多的表面活性位点,使其循环次数提升至10次以上.Abstract: FeOCl serves as an effective heterogeneous Fenton catalyst, demonstrating excellent catalytic performance across a wide pH range; however, its pH adaptability is significantly influenced by water quality. This study prepared a partially pyrolyzed FeOCl catalyst and investigated the buffering effect of wastewater pH on total organic carbon (TOC) mineralization during the FeOCl Fenton reaction, utilizing leather biochemical tailwater as the target. The behavior was also compared under conditions using the model compound gallic acid (GA). Results indicate that under optimal reaction conditions (6 mmol·L−1 H2O2, 3.5 g·L−1 FeOCl), FeOCl can generate approximately 7.6 × 10−4 mol·L−1 of H+ through hydrolysis and the Fenton process, rapidly reducing the initial pH from neutral to between 3 and 4, at which point the TOC mineralization rate reaches approximately 70%. For actual wastewater, FeOCl can introduce about 3.75 × 10−3 mol·L−1 of H+ under operational conditions, necessitating only 2.25 × 10−3 mol·L−1 of additional acid to adjust the pH to 4, achieving mineralization effects similar to those under optimal conventional Fenton conditions at an initial pH of 6. The gradual deactivation of the catalyst is primarily attributed to the physical adsorption of organics and the complexation of Fe3+ with carboxyl groups in wastewater, which reduces active Fe sites. However, acid washing combined with ultrasonic treatment effectively desorbs organics, exposing more surface active sites and enhancing recycling capacity to over 10 cycles.
-
Key words:
- FeOCl /
- heterogeneous Fenton /
- pH buffering /
- advanced oxidation /
- wastewater treatment.
-

-

图 1 不同升温速率下制备的FeOCl电镜图
Figure 1. SEM images of FeOCl prepared at different heating rates

图 2 不同升温速率下制备的FeOCl的XRD谱(a)及其对GA溶液中TOC去除效果(b)
Figure 2. XRD images and TOC removal effect of FeOCl prepared at different heating rates XRD(a), TOC removal effect (b)

图 4 不同FeOCl与H2O2投加量对催化效果的影响
Figure 4. The effect of different FeOCl and H2O2 dosages on catalytic performance

图 5 (a)不同没食子酸浓度体系的EPR谱图,(b)不同没食子酸浓度体系5 min时的·OH产量以及40 min内的H2O2消耗量,(c、d) 20 mg·L−1和80 mg·L−1 GA体系反应后FeOCl表面Fe元素XPS
Figure 5. (a) EPR spectra of systems with different GA concentrations,(b) ·OH yield at 5 min and H2O2 consumption over 40 min for systems with different gallic acid concentrations, (c,d) XPS of elemental Fe on the surface of FeOCl after reaction with 20 mg·L−1 and 80 mg·L−1 GA systems

图 6 100 mL模拟废水与实际废水的酸缓冲滴定曲线
Figure 6. Acid buffer titration curves for 100 mL of simulated and actual wastewater

图 7 不同FeOCl投加顺序对TOC矿化效果及体系pH的影响
Figure 7. Effects of different FeOCl dosing sequences on the mineralization effect of TOC and pH of the system

图 8 不同初始pH值条件下不同体系对实际废水的TOC去除效果及体系pH变化趋势
Figure 8. TOC removal effects of different systems on actual wastewater under different initial pH values and the changing trend of system pH
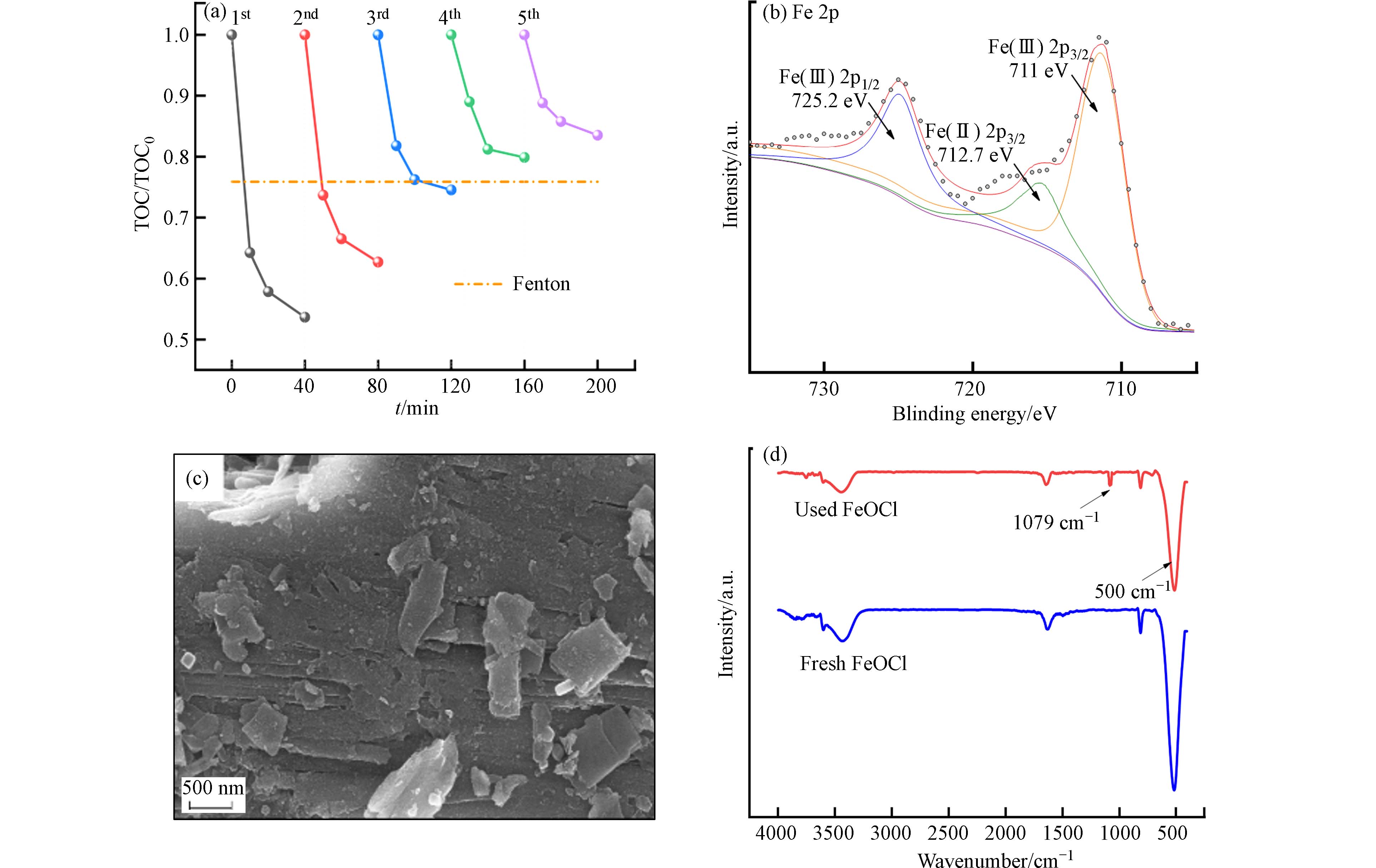
图 9 (a)FeOCl直接循环5次TOC矿化效果,(b)循环5次后FeOCl表面Fe元素XPS,(c)5次循环后的FeOCl表面SEM,(d)5次反应前后红外光谱图
Figure 9. (a) TOC mineralization effect of FeOCl direct cycling for five times, (b) XPS diagram of Fe elements on the catalyst surface after five cycles, (c) SEM diagram of the catalyst surface after five cycles, (d) infrared spectra before and after five reactions
-
[1] YAN Q Y, LIAN C, HUANG K, et al. Constructing an acidic microenvironment by MoS2 in heterogeneous Fenton reaction for pollutant control[J]. Angewandte Chemie (International Ed), 2021, 60(31): 17155-17163. doi: 10.1002/anie.202105736 [2] GAO P, REDDY M A, MU X K, et al. VOCl as a cathode for rechargeable chloride ion batteries[J]. Angewandte Chemie (International Ed), 2016, 55(13): 4285-4290. doi: 10.1002/anie.201509564 [3] JI X X, WANG H F, HU P J. First principles study of Fenton reaction catalyzed by FeOCl: Reaction mechanism and location of active site[J]. Rare Metals, 2019, 38(8): 783-792. doi: 10.1007/s12598-018-1140-9 [4] JARRIGE I, CAI Y Q, SHIEH S R, et al. Charge transfer in FeOCl intercalation compounds and its pressure dependence: An X-ray spectroscopic study[J]. Physical Review B, 2010, 82(16): 165121. doi: 10.1103/PhysRevB.82.165121 [5] 王金岭, 温玉真, 汪华林, 等. FeOCl层状材料及其插层化合物: 结构、性质与应用[J]. 化学进展, 2021, 33(2): 263-280. WANG J L, WEN Y Z, WANG H L, et al. FeOCl layered materials and intercalated compounds: Structure, properties and applications[J]. Advances in Chemistry, 2021, 33(2): 263-280 (in Chinese).
[6] ZHAO X Y, ZHANG Z H. FeOCl in advanced oxidization processes for water purification: A critical review[J]. Current Pollution Reports, 2023, 9(2): 143-164. doi: 10.1007/s40726-023-00256-9 [7] CAO Y, CUI K P, CHEN Y H, et al. Efficient degradation of tetracycline by H2O2 catalyzed by FeOCl: A wide range of pH values from 3 to 7[J]. Solid State Sciences, 2021, 113: 106548. doi: 10.1016/j.solidstatesciences.2021.106548 [8] 马金环, 魏智强, 赵继威, 等. FeOCl光芬顿催化剂的表征及其降解罗丹明B的效果[J]. 材料科学与工艺, 2023, 31(6): 9-18. MA J H, WEI Z Q, ZHAO J W, et al. Characterization of FeOCl photo-Fenton catalyst and its degradation effect on Rhodamine B[J]. Materials Science and Technology, 2023, 31(6): 9-18 (in Chinese).
[9] 张少朋, 陈瑀, 白淑琴, 等. 氯氧铁非均相催化过氧化氢降解罗丹明B[J]. 环境科学, 2019, 40(11): 5009-5014. ZHANG S P, CHEN Y, BAI S Q, et al. Catalytic degradation of rhodamine B by FeOCl activated hydrogen peroxide[J]. Environmental Science, 2019, 40(11): 5009-5014 (in Chinese).
[10] 王晴. 制革复鞣废水中聚合物-Cr的络合及去除行为研究[D]. 西安: 陕西科技大学, 2020. WANG Q. Study on complexation and removal behavior of polymer-Cr in tanning wastewater[D]. Xi’an: Shaanxi University of Science & Technology, 2020 (in Chinese).
[11] 高明明, 柴晓苇, 曾运航, 等. 制革废水中的氯离子含量及来源分析[J]. 皮革科学与工程, 2013, 23(5): 46-50. GAO M M, CHAI X W, ZENG Y H, et al. Chloride ion content in wastewaters of leather making processes and analysis of its origin[J]. Leather Science and Engineering, 2013, 23(5): 46-50 (in Chinese).
[12] 孙柏阳, 马宏瑞, 朱超, 等. 铬屑碱解过程中铬与胶原分子量分布特征[J]. 环境化学, 2022, 41(10): 3447-3456. doi: 10.7524/j.issn.0254-6108.2022051802 SUN B Y, MA H R, ZHU C, et al. Dechromation and molecular weight distribution of hydrolyzed collagen from chromium-containing leather during alkaline[J]. Environmental Chemistry, 2022, 41(10): 3447-3456 (in Chinese). doi: 10.7524/j.issn.0254-6108.2022051802
[13] 冯岱. 黄牛鞋面革浸水工艺控制要点[J]. 中国皮革, 2020, 49(1): 58-59. FENG D. Key points of soaking process for cattle upper leather[J]. China Leather, 2020, 49(1): 58-59 (in Chinese).
[14] 顾家熙, 马宏瑞, 李晓洁, 等. 制革生化尾水Fenton法处理对铬存在形态的影响[J]. 中国皮革, 2019, 48(1): 53-58. GU J X, MA H R, LI X J, et al. Effect of Fenton method on existence of chromium in tannery secondary effluent[J]. China Leather, 2019, 48(1): 53-58 (in Chinese).
[15] 杨应秋, 魏杰. 没食子酸加速卡马西平在Fe(Ⅲ)/PDS中的降解[J]. 工业水处理, 2021, 41(12): 95-101. YANG Y Q, WEI J. Degradation of carbamazepine in Fe(Ⅲ)/PDS system under Gallic acid acceleration[J]. Industrial Water Treatment, 2021, 41(12): 95-101 (in Chinese).
[16] 侯晓静. 异相Fenton体系铁循环调控及其污染物降解性能增强[D]. 武汉: 华中师范大学, 2018. HOU X J. Regulation of iron cycle in heterogeneous Fenton system and enhancement of pollutant degradation performance[D]. Wuhan: Central China Normal University, 2018 (in Chinese).
[17] 黄腾腾. FeOCl光催化材料的制备改性及其处理RhB废水的研究[D]. 西安: 长安大学, 2021. HUANG T T. Preparation and modification of FeOCl photocatalytic material and its treatment of RhB wastewater[D]. Xi’an: Changan University, 2021 (in Chinese).
[18] 曹勇. FeOCl及其改性材料非均相催化降解盐酸四环素和诺氟沙星的研究[D]. 合肥: 合肥工业大学, 2021. CAO Y. Heterogeneous catalytic degradation of tetracycline hydrochloride and norfloxacin by FeOCl and its modified materials[D]. Hefei: Hefei University of Technology, 2021 (in Chinese).
[19] ZHU G P, YU X D, XIE F, et al. Ultraviolet light assisted heterogeneous Fenton degradation of tetracycline based on polyhedral Fe3O4 nanoparticles with exposed high-energy{110}facets[J]. Applied Surface Science, 2019, 485: 496-505. doi: 10.1016/j.apsusc.2019.04.239 [20] HOU L W, WANG L G, ROYER S, et al. Ultrasound-assisted heterogeneous Fenton-like degradation of tetracycline over a magnetite catalyst[J]. Journal of Hazardous Materials, 2016, 302: 458-467. doi: 10.1016/j.jhazmat.2015.09.033 [21] CHEN M D, XU H M, WANG Q, et al. Activation mechanism of sodium percarbonate by FeOCl under visible-light-enhanced catalytic oxidation[J]. Chemical Physics Letters, 2018, 706: 415-420. doi: 10.1016/j.cplett.2018.06.004 [22] NEYENS E, BAEYENS J. A review of classic Fenton’s peroxidation as an advanced oxidation technique[J]. Journal of Hazardous Materials, 2003, 98(1/2/3): 33-50. [23] SUN M, CHU C H, GENG F L, et al. Reinventing Fenton chemistry: Iron oxychloride nanosheet for pH-insensitive H2O2 activation[J]. Environmental Science & Technology Letters, 2018, 5(3): 186-191. [24] YANG X J, XU X M, XU J, et al. Iron oxychloride (FeOCl): An efficient Fenton-like catalyst for producing hydroxyl radicals in degradation of organic contaminants[J]. Journal of the American Chemical Society, 2013, 135(43): 16058-16061. doi: 10.1021/ja409130c [25] PLIEGO G, ZAZO J A, GARCIA-MUÑOZ P, et al. Trends in the intensification of the Fenton process for wastewater treatment: An overview[J]. Critical Reviews in Environmental Science and Technology, 2015, 45(24): 2611-2692. doi: 10.1080/10643389.2015.1025646 [26] 张亚珍. 过氧化氢的漂白及其稳定性探讨[J]. 天津纺织工学院学报, 1988, 7(1): 105-109. ZHANG Y Z. Hydrogen peroxide bleaching and its stability[J]. Journal of Tiangong University, 1988, 7(1): 105-109 (in Chinese).
[27] NAVALON S, ALVARO M, GARCIA H. Heterogeneous Fenton catalysts based on clays, silicas and zeolites[J]. Applied Catalysis B: Environmental, 2010, 99(1/2): 1-26. -
 点击查看大图
点击查看大图
计量
- 文章访问数: 273
- HTML全文浏览数: 273
- PDF下载数: 13
- 施引文献: 0
