

-
甲醛(HCHO)是一类常见的室内空气污染物,主要来自建筑材料、家具和各种装饰材料[1-3]。2000年,世界卫生组织提出长时间接触甲醛的限值不应超过0.2 mg·m−3[4]。长期暴露于超过安全限制的HCHO可能会引发严重的健康问题,比如眼睛、皮肤喉咙发炎以及咳嗽、支气管炎,甚至鼻咽癌和白血病[5-6]。因此,有效清除甲醛对于改善空气质量和保护健康非常重要。各种去除HCHO的方法已在文献中报道,包括物理或化学吸附[7-8],等离子体技术[9-10],光催化氧化[11]和催化氧化(热和非热)。室温下催化氧化HCHO被认为是最有前途的技术,可以完全转化为CO2和H2O,不会造成二次污染。
用于室温催化氧化HCHO的催化材料可分为两类∶负载贵金属(Pt、Pd、Au和Ag)和非贵金属氧化物(MnO2、CuO、TiO2等)[12]。负载贵金属催化剂在室温下具有较高的催化氧化HCHO为CO2和H2O,在ZHANG等[13]的研究中,二氧化钛负载铂在室温下氧化HCHO具有优异的催化性能;然而,贵金属催化剂由于成本高、资源有限和热稳定性差,在实际应用中存在局限性[14]。为了取代贵金属催化剂,研究人员的目标之一是寻找催化活性高、稳定性好的非贵金属催化剂。非贵金属多为过渡金属,含有不饱和的d轨道,容易吸附反应物分子并活化,具有很好的催化活性[15-16]。Mn、Co、Ce、Cu等过渡金属的氧化物因具有多种化合价态,经过预处理后会在催化剂表面产生缺陷进而增强吸附和活化VOCs的能力。由于具有独特的晶体结构、高催化活性和低成本,二氧化锰是最有前途的甲醛催化剂之一[17]。氧化锆因其高腐蚀性,高机械和热阻以及优异的氧迁移率成为一种有吸引力的过渡金属氧化物[18-19]。单一锰氧化物在催化VOCs活性不理想,引入第二金属组分Zr来对单一锰氧化物进行改性,使之形成锰锆氧化物催化剂。DENG等[20]采用溶胶-凝胶法制备的Mn0.5Zr0.5催化氯乙烯转化率可达96%;焦坤灵等[21]采用溶胶凝胶法制备Mn/Zr负载稀土尾矿的NH3-SCR催化剂,其脱硝活性可达96%;谈冠希等[22]采用柠檬酸络合法制备锰锆复合氧化物催化剂与纯相MnOx相比,CO催化还原NO活性有所提升。基于此,将ZrO2掺杂到单一锰氧化物中,能够诱导更高的晶格氧迁移率和高反应活性[23]。但使用锰锆氧化物去除甲醛方面的研究不多。
本研究采用简单的共沉淀法来制备MnxZr1-xOy催化剂,通过考察不同Mn与Zr摩尔比对室温催化氧化甲醛的影响,筛选出催化氧化甲醛性能最佳的催化剂。结合各种表征手段,分析MnxZr1-xOy催化剂的催化性能与其物理化学结构间的关系。
-
所有用于制备的化学品均为分析级,水为去离子水,催化剂共沉淀法制备。将适量的硝酸锆五水合物和硝酸锰(Mn∶Zr(摩尔比)=0.33、1、2、4)在100 ml去离子水中搅拌溶解至澄清透明,加入氨水调节pH至9,搅拌3 h,所得沉淀抽滤并用去离子水洗涤3次,80 ℃干燥12 h,最后以5 ℃·min−1、500 ℃、4 h焙烧,研磨过筛(40~60目)得到催化剂样品,记为MnxZr1-xOy(Mn1.0Zr0Oy、Mn0.8Zr0.2Oy、Mn0.67Zr0.33Oy、Mn0.5Zr0.5Oy和Mn0.25Zr0.75Oy催化剂)。
-
采用扫描电镜(FEI, Nova Nano SEM450, USA)观察催化剂的微观形貌。用透射电镜分析催化剂的晶格间距,以乙醇为分散剂,用普通铜网进行样品制备。在CuKα、2θ=10º~80º、扫描速率10 º·min−1、步长0.01 º·s−1的条件下,采用德国Bruker D8 Advance X射线衍射仪测定样品的相组成。用Mack
2460 分析仪测定了催化剂的比表面积和孔径等孔隙结构参数。X射线光电子能谱采用Thermo Scientific NEXSA, Al Kα进行表征,工作电压为15 kV,结合能采用内标C1s峰(Eb=284.80 eV)标定,精度为±0.2 eV。用傅立叶红外光谱(Thermo Scientific, Nicolet Is20, USA)分析催化剂的分子结构。采用氧气程序升温脱附(O2-TPD)分析催化剂内部活性氧的位点。 -
采用自制装置评价催化剂活性,在常压石英管(直径为6 mm)固定床反应器中对HCHO进行催化氧化试验。100 mg的催化剂装入反应器中,引入含1.5 mg·m−3HCHO的模拟气流,载气为N2+20%O2,气体总流量100 mL·min−1。用酚试剂分光光度法分析反应物或产物气流中的HCHO浓度。将含有微量HCHO的气流通过5 mL酚试剂溶液起泡30 min,通过吸收收集HCHO。然后加入0.4 mL (质量分数为1%)硫酸铁铵溶液作为着色剂,振荡5 s,静置15 min后,用分光光度计测量630 nm光吸光度,测定气流中HCHO浓度。根据HCHO的浓度变化计算其转化率,HCHO转化率按式(1)计算。
式中∶XHCHO为甲醛的去除率,%;Cin为甲醛的进口浓度,mg·m−3;Cout为甲醛的出口浓度,mg·m−3。
为了确定实验产物,采用体积滴定法测定反应产物中二氧化碳的浓度,过量的氢氧化钡溶液与产物中的二氧化碳反应生成碳酸钡沉淀。反应结束后,用标准草酸溶液滴定剩余的氢氧化钡,直至酚酞的红色刚好褪去,反应后的二氧化碳浓度可以通过体积滴定(式(2))计算。
式中∶φ为空气中二氧化碳体积分数,%;V1为样品滴定所用草酸标准溶液体积,单位mL;V2为空白滴定所用草酸标准溶液体积,单位mL;V0为标准状态下的采气体积,单位为L。
-
图1(a)为不同Mn/Zr摩尔比的MnxZr1-xOy催化剂的XRD测定结果。由图1(a)可以看到,锰锆摩尔比对催化剂的结晶度有显著影响,当x=1.0(只含Mn不含Zr)时,只出现了Mn3O4(PDF#80-0382)的特征衍射峰,分别位于2θ为18.01°、28.91°、32.38°、36.08°、38.09°、44.04°、50.83°、59.90°、64.01°。当x=0.5,0.67,0.8时,未检测到Zr物种相关的特征峰,只出现了Mn2O3(PDF#71-0635)的特征峰,而Mn2O3的特征峰向低角度方向偏移,并且衍射峰强度变弱、峰形宽化。可能是因为较小的Mn3+离子半径(0.67 Å)被较大的Zr4+离子半径(0.79 Å)替代[23],从而渗进到Mn2O3晶格中,形成锰锆固溶体结构,这有利于锰锆之间的相互协同作用,促使其对甲醛的催化活性显著提高。当x=0.25时,出现Mn0.2Zr0.8O1.8 (PDF#77-
2157 )的特征衍射峰,这表明Mn和Zr形成了Mn0.2Zr0.8O1.8固溶体,且衍射峰强度高,峰形尖锐。图1(b)为不同Mn∶Zr摩尔比的MnxZr1-xOy催化剂的FT-IR测定结果。可见,Mn0.8Zr0.2Oy、Mn0.67Zr0.33Oy、Mn0.5Zr0.5Oy、Mn0.25Zr0.75Oy催化剂的FT-IR图谱基本一致,且吸收峰位于同一波数处,说明不同的锰锆摩尔比对样品的结构影响不大。3 424 cm−1的峰被认为是游离的—OH伸缩振动峰[24];1 384 cm−1处的峰被认为是硝酸盐中v(N—O)的不对称伸缩振动,表明样品中可能存在少量的含氮物质[25];632、526、436 cm−1被认为是Mn—O键的伸缩振动[26]。当x=1时当x=1时,存在Mn—O键的吸收峰,XRD的测试结果也表明主要物相是Mn3O4,进一步证明存在Mn—O键;当x<1时,由于锰和锆之间的协同作用未能见到Mn—O键的吸收峰,提高了锰锆氧化物催化氧化甲醛的效率。
-
为了观察催化剂的表面形貌,对催化剂进行了SEM表征分析。由图2(a)可以看出, Mn0.67Zr0.33Oy催化剂的微观形貌呈现不规则的块状,可能是由于共沉淀制备样品所致。为了进一步观察Mn0.67Zr0.33Oy催化剂的晶格条纹及内部精细结构,进行TEM分析,由图2(b)、(d)可知,样品整体为规则晶体与不规则无定型结构的复合形貌,高分辨下规则形貌的晶格条纹明显,不规则区域基本为无定型结构。利用Gatan digital micrograph 从TEM图像计算晶格间距,得到Mn2O3 (040)和Mn2O3 (412)平面的晶格间距(d)分别为0.212 2 nm和0.243 5 nm,如图2(e)所示,这与XRD检测的主要物相为Mn2O3一致。此外,利用TEM HADDF对Mn0.67Zr0.33Oy催化剂进行元素映射分析(图2(f)),进一步观察复合材料的空间分布和组合。结果表明,Mn、Zr、O在Mn0.67Zr0.33Oy催化剂中分布良好,EDS-mapping显示规则晶体处Mn元素集中分布,不规则形貌处Zr元素集中分布。
-
图3(a)为各催化剂的N2吸脱附等温线。由图3(a)可知,Mn1.0Zr0Oy催化剂该等温线属IUPAC分类中III型,相对压力较低时,为单分子层吸附。Mn0.25Zr0.75Oy、Mn0.5Zr0.5Oy、Mn0.67Zr0.33Oy和Mn0.8Zr0.2Oy的吸脱附等温线具有相同形状,属于典型的IUPAC分类中IV型等温线,在P/P0为0.4~1.0内检测到H4型滞回,表明存在狭缝状的介孔结构。从图中可以看出,在低压段吸附量平缓增加,此时N2分子以单层到多层吸附在介孔的内表面。图3(b)为相应的BJH孔径分布,Mn0.25Zr0.75Oy、Mn0.5Zr0.5Oy、Mn0.67Zr0.33Oy和Mn0.8Zr0.2Oy催化剂的孔径分别为3.78、3.4 、3.8和3.8 nm,说明样品为介孔结构,有利于反应物的传输;而Mn1.0Zr0Oy催化剂的孔径主要分布在2.4 nm和49.2 nm,为双峰孔径分布。
MnxZr1-xOy催化剂的比表面积、孔容和孔径如表1所示。由表1可以看出,制备的单组分Mn1.0Zr0Oy催化剂的比表面积只有15.3 m²·g−1,然而制备的锰锆氧化物的比表面积为105.2~215.1 m²·g−1,大约为Mn1.0Zr0Oy催化剂的7~14倍,其中锰锆摩尔比为2∶1时,得到Mn0.67Zr0.33Oy催化剂具有最大的比表面积(215.1 m²·g−1),为反应物充分接触催化剂活性位点提供了较大的反应界面,从而使得催化剂拥有优异的催化氧化甲醛活性。Zr的添加使得催化剂的比表面积先增大后减小,这表明锰锆之间的相互作用可以大幅度提高MnxZr1-xOy催化剂的比表面积,这可能是因为锆氧化物本身具有很强的稳定性,与锰相互作用抑制了锰氧化物在制备条件下的团聚。这表明适当的掺杂可以提高催化剂的比表面积,且较高的比表面积在催化氧化甲醛方面效果明显提升,相较于单组份Mn1.0Zr0Oy催化剂在6 h甲醛的去除率只能保持在62%,且稳定性并不好;而Mn0.67Zr0.33Oy催化剂的甲醛去除率在6 h依旧保持在100%,在后续的稳定性测试中,甲醛的去除率保持在90%以上可达50 h,延长了催化剂的使用寿命。
-
为了进一步确定催化剂表面的元素价态变化,对催化剂进行了XPS分析。图4为Mn2p、O1s和Zr3d相关的峰,表2是对各催化剂的XPS图谱进行积分处理后得到的数据。由图4(a)可知,各催化剂的XPS光谱图中含有Mn、Zr、O特征峰,3种元素共存且不含其他杂质峰,而Mn1.0Zr0Oy催化剂未能见到Zr的峰。图4(b)为不同MnxZr1-xOy催化剂的Mn2p图谱,Mn2p1/2是结合能约为654 eV时的峰值和Mn2p3/2是结合能约为642 eV时的峰值[27-28],Mn2p3/2分为位于641.4、642.5、644.5 eV的3个峰,分别对应Mn2+、Mn3+、Mn4+物种。人们普遍认为高浓度的表面Mn4+有利于通过可逆氧化还原循环低温氧化HCHO[29]。由表2可知,Mn0.67Zr0.33Oy催化剂中Mn4+含量为15.75%,Mn3+含量为44.39%,反应后Mn4+含量降至11.51%,Mn3+含量升至54.57%。这表明Mn4+被还原为Mn3+,表面Mn4+含量降低,不利于氧化甲醛,致使催化氧化甲醛的活性下降。而Mn1.0Zr0Oy催化剂中Mn4+含量只有6.85%,在室温下对甲醛的去除效率不高。
图4(c)为O1s的XPS图。结合能在529.7~530.3、531.4~531.8和532.9~533.7 eV的特征峰分别对应于氧化物的晶格氧(Olatt)、表面活性氧(Oads)和表面吸附的碳酸盐或水(OH2O)[30-32]。现有研究[33]表明,表明活性氧对污染物的催化分解至关重要。表面活性氧如O2−、O−和-OH具有较强的氧化能力,因此,使用O1s的 XPS图谱对表面活性氧进行了评价。从表2可知,Mn0.67Zr0.33Oy的表面活性氧含量为0.34,其含量在所有催化剂中最高,在催化氧化甲醛的反应中保持较高的活性且较好的稳定性。随着HCHO反应的继续,表面活性氧和晶格氧的含量下降,表面吸附的碳酸盐和水反而增加,这表明表面活性氧在催化氧化甲醛的反应中起着重要作用。Mn1.0Zr0Oy催化剂中表面活性氧的含量最低(仅为0.21),而晶格氧含量最高(0.65),表明晶格氧不利于HCHO的催化氧化,甲醛的去除效果不佳。由此可以看出,掺杂锆元素后的催化剂表面吸附氧含量明显增加。当锰与锆摩尔比为2∶1时,催化剂表面活性氧含量最高,继续增加锰的含量,表面活性氧的含量反而有所下降。
图4(d)为Zr3d的XPS图谱。中心峰位于182 eV,肩峰位于184.3 eV,与ZrO2和Zr4+的特征峰一致,并未出现明显的偏移,表明锰锆摩尔比不会影响锆的峰位置,Mn1.0Zr0Oy催化剂并未出现Zr的特征峰,这与制备条件一致。
-
Mn0.67Zr0.33Oy催化剂反应前后的O2-TPD图如图5所示。一般来说,在低温条件下的脱附峰是催化剂表面上化学吸附氧的脱附和吸附在氧空位上的氧的脱附,而在较高温度下的脱附峰是样品中晶格氧逸出所致[34]。因此,表面活性氧主要表现在134 ℃、139 ℃时较低的氧解吸峰值,而晶格氧解吸峰一般表现在555、574、679 ℃附近的氧解吸峰。通过对比反应前后的O2-TPD图,可以发现,反应后的脱附峰向高温移动,且氧脱附峰信号减弱。这表明表面活性氧降低,导致催化氧化甲醛的效率下降。
-
室温下MnxZr1-xOy催化剂摩尔比对甲醛去除率的影响结果如图6(a)所示。总体而言,催化剂的活性排序为Mn0.67Zr0.33Oy>Mn0.8Zr0.2Oy>Mn0.5Zr0.5Oy>Mn0.25Zr0.75Oy>Mn1.0Zr0Oy。Mn1.0Zr0Oy催化剂由于未掺杂Zr元素,甲醛的去除率在6 h内仅为61%,12 h后急剧下降到29%,去除效果差。而掺杂Zr元素之后,催化剂催化氧化甲醛的效率大大提升,其中Mn0.67Zr0.33Oy表现出优异的甲醛去除率,6 h内活性达到100%,反应12 h后依旧保持在97%,24 h为96.72%,在后续的反应中,该催化剂去除甲醛的效果在90%以上可持续50 h,如图6(c)所示,高于其他催化剂的最佳甲醛去除率。QIAN等[35]采用溶胶-凝胶制备锰铈复合氧化物,室温下24 h的甲醛去除率为90.9%;而本研究所制备的Mn0.67Zr0.33Oy催化剂室温下24 h的甲醛去除率为96%。周辉等[36]使用氧化还原法制备二氧化锰催化剂,对甲醛的催化降解活性在300 min以内保持在80%以上,而本研究制备的Mn0.67Zr0.33Oy催化剂300 min以内可以100%降解甲醛。Mn0.8Zr0.2Oy、Mn0.5Zr0.5Oy和Mn0.25Zr0.75Oy样品在300 min内对甲醛的去除率分别为97.8%、91.4%和87%,且随着反应时间的延长,HCHO去除率呈现下降趋势。
BET表征结果表明,Mn0.67Zr0.33Oy催化剂拥有最大的比表面积(215.1 m²·g−1),为反应物充分接触催化剂活性位点提供了较大的反应界面;XPS表征结果表明,Mn0.67Zr0.33Oy催化剂表面拥有最高的表面活性氧(0.34),促进了甲醛的催化氧化,反应后的表面活性氧含量下降,Mn4+的含量降低,Mn3+的含量增加,Mn4+被还原为Mn3+,导致催化氧化甲醛的性能下降。
室温下Mn与Zr摩尔比对CO2选择性的影响结果如图6(b)所示。如图6(b)所示,Mn/Zr摩尔比对CO2的选择性有显著的影响。Mn0.67Zr0.33Oy催化剂的CO2选择性在6 h内为100%,说明Mn0.67Zr0.33Oy催化剂能够有效地将HCHO氧化为CO2,进一步说明了该反应为催化氧化。为了解Mn0.67Zr0.33Oy在反应过程中是否稳定,对反应前后的样品进行FT-IR表征,结果如图7所示。可见,反应前后未出现新的峰,说明反应过后并未出现新的官能团,表明Mn0.67Zr0.33Oy在反应过程中比较稳定。
-
1)通过简单的共沉淀法制备了MnxZr1-xOy催化剂,MnxZr1-xOy可实现100%去除甲醛,且具有良好的稳定性,能够长时间去除甲醛(50 h后甲醛的去除率依旧在90%以上)。
2)当Mn/Zr摩尔比为2∶1时,催化剂的性能最佳,Mn0.67Zr0.33Oy催化剂比表面积最大(215.1 m²·g−1),较高的比表面积有利于暴露更多的活性位点,为反应物充分接触催化剂活性位点提供了较大的反应界面∶表面活性氧含量最高(0.34),Mn4+含量高,更多的活性氧有利于吸附氧的活化,这对甲醛的催化氧化起着关键作用。
高比表面积MnxZr1-xOy催化剂的制备及其在室温下催化氧化甲醛的效果
Preparation of high specific surface area MnxZr1-xOy catalyst and its catalytic oxidation effect of formaldehyde at room temperature
-
摘要: 甲醛作为室内空气污染物之一,对人体产生的危害不可逆转。因此,高效去除甲醛成为研究热点之一。该研究采用共沉淀法得到不同活性的MnxZr1-xOy催化剂,利用XRD、FT-IR、SEM、TEM、BET、XPS、O2-TPD等方法对材料进行了表征分析,评价了不同锰锆摩尔比在室温下催化氧化甲醛的影响效果。结果表明,锰与锆的摩尔比为2∶1时反应得到的Mn0.67Zr0.33Oy催化氧化甲醛效果最好,去除率达到100%,反应50 h后催化剂的活性仍在90%以上。表征结果表明,Mn0.67Zr0.33Oy催化剂拥有最大的比表面积(215.1 m²·g−1),加大了气体与催化剂的接触面积,表面活性氧和高价态Mn含量高成为催化剂性能优异的主要原因。
-
关键词:
- MnxZr1-xOy /
- 甲醛 /
- 催化氧化 /
- 室温
Abstract: Formaldehyde, as one of the indoor air pollutants, produces irreversible harm to the human body. Therefore, the removal efficient of formaldehyde has become one of the research hotspots. In this study, MnxZr1-xOy catalysts with different activities were obtained by the co-precipitation method, and the materials were characterized by XRD, FT-IR, SEM, TEM, BET, XPS and O2-TPD. Effect of different manganese-zirconium molar ratios for the catalytic oxidation of formaldehyde at room temperature were evaluated. The results showed that catalytic oxidation effect of formaldehyde by Mn0.67Zr0.33Oy obtained from manganese and zirconium at a molar ratio of 2∶1 was the best, with the removal rate of formaldehyde reaching 100%, and the catalytic activity was above 90% after 50 h of reaction. The characterization results showed that the Mn0.67Zr0.33Oy catalyst possessed the largest specific surface area (215.1 m²·g−1), which increases the contact area between the gas and the catalyst, and high surface-activated oxygen and high valence Mn content are the main reasons for the excellent performance of the catalysts.-
Key words:
- MnxZr1-xOy /
- formaldehyde /
- catalytic oxidation /
- room temperature
-

-
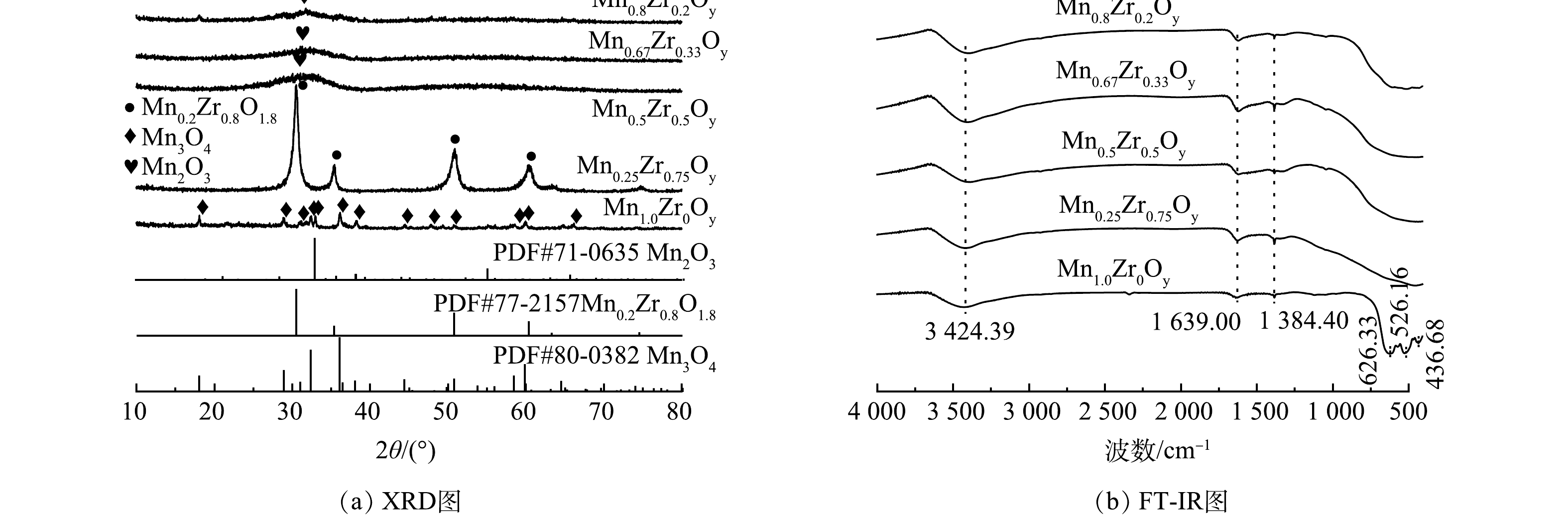
图 1 不同Mn/Zr摩尔比的MnxZr1-xOy催化剂的XRD图和FTIR图
Figure 1. XRD pattern and FTIR pattern of MnxZr1-xOy catalyst with different Mn/Zr molar ratios

图 2 Mn0.67Zr0.33Oy催化剂的形貌表征
Figure 2. Morphological characterization of Mn0.67Zr0.33Oy catalysts

图 3 不同Mn/Zr摩尔比催化剂的N2吸附-脱附曲线和孔径分布
Figure 3. N2 adsorption-desorption curves and pore size distribution of catalysts with different Mn/Zr molar ratios

图 4 不同Mn/Zr摩尔比的MnxZr1-xOy催化剂的XPS图
Figure 4. XPS spectra of MnxZr1-xOy catalysts with different Mn/Zr molar ratios

图 5 Mn0.67Zr0.33Oy催化剂反应前后的O2-TPD曲线图
Figure 5. O2-TPD profiles before and after the reaction of Mn0.67Zr0.33Oy catalysts
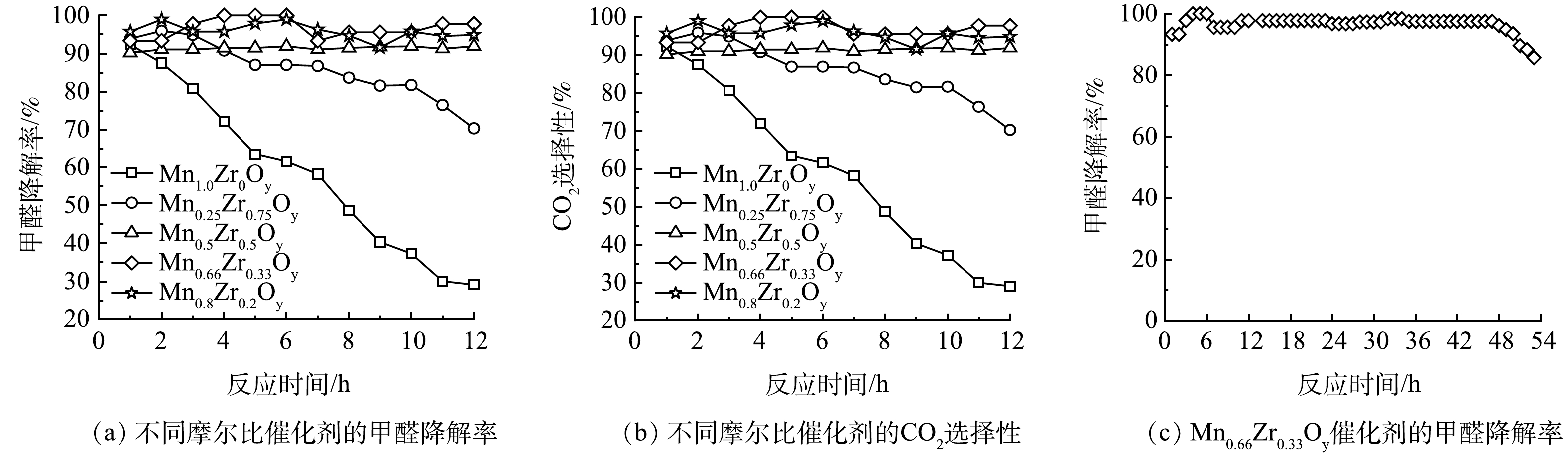
图 6 不同摩尔比催化剂的性能分析
Figure 6. Performance analysis of catalysts with different molar ratios
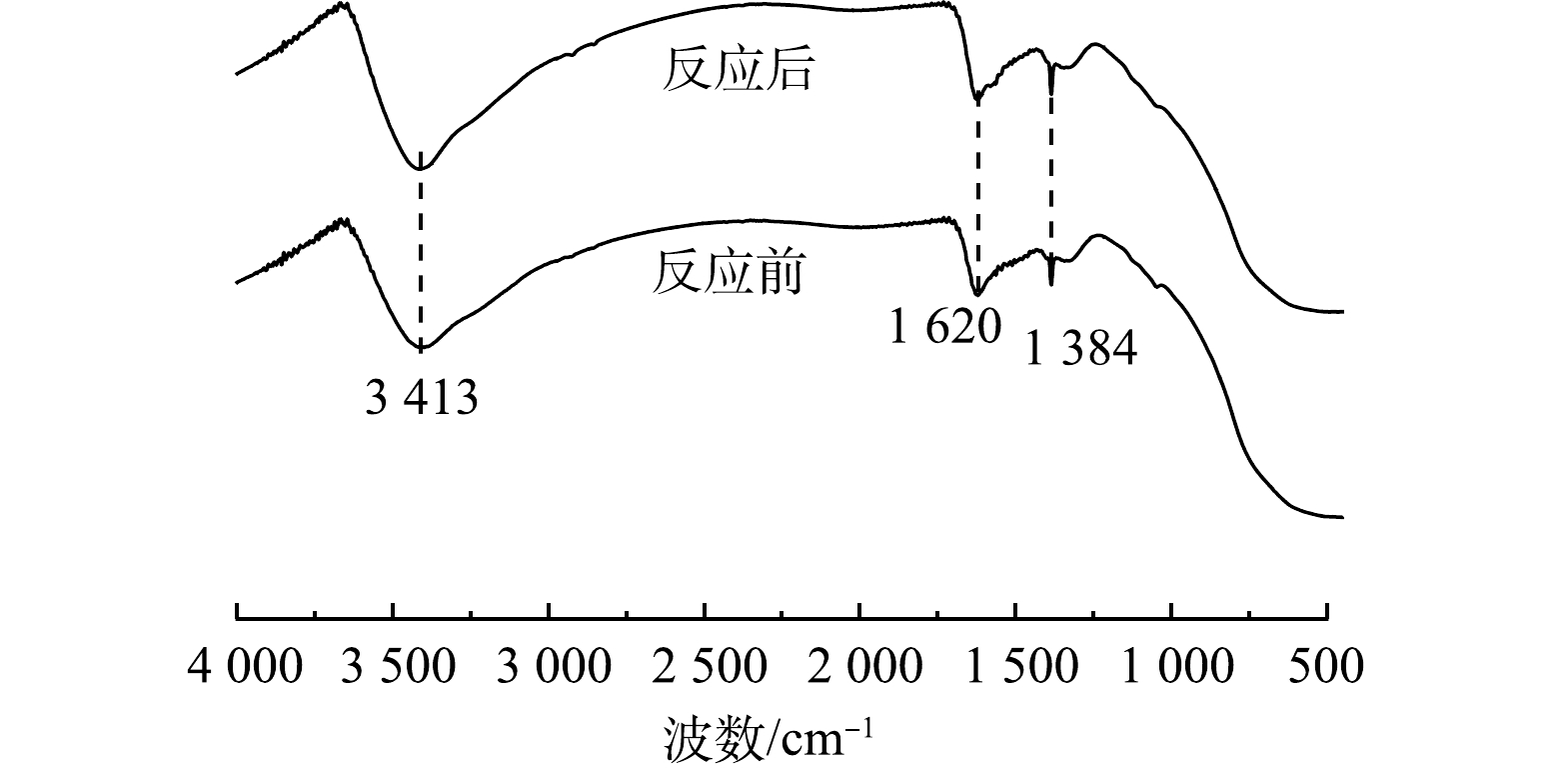
图 7 Mn0.66Zr0.33Oy催化剂反应前后的FTIR图谱
Figure 7. FTIR pattern of Mn0.66Zr0.33Oy catalyst before and after reaction
表 1 MnxZr1-xOy催化剂的比表面积、孔容和孔径
Table 1. Specific surface area, pore volume and pore size of the MnxZr1-xOy catalyst
样品 比表面积/(m²·g−1) 总孔容/(cm³·g−1) 平均孔径/nm Mn1.0Zr0Oy 15.3 0.096 28.083 Mn0.25Zr0.75Oy 105.2 0.149 4.579 Mn0.5Zr0.5Oy 162.9 0.166 5.005 Mn0.67Zr0.33Oy 215.1 0.269 4.909 Mn0.8Zr0.2Oy 184.6 0.288 6.194  下载: 导出CSV
下载: 导出CSV
表 2 MnxZr1-xOy催化剂Mn2p、O1s峰曲线拟合结果
Table 2. Mn2p, O1s peak curve fitting results for MnxZr1-xOy catalyst
样品 元素比 Mn2+ Mn3+ Mn4+ Mn3+/Mn4+ Oads/(Oads+
Olatt+OH2O)/%Mn1.0Zr0Oy 75.1 18.05 6.85 2.63 0.21 Mn0.25Zr0.75Oy 50.09 40.77 14.78 2.76 0.28 Mn0.5Zr0.5Oy 31.52 53.5 14.98 3.57 0.27 Mn0.67Zr0.33Oy-反应前 39.87 44.39 15.75 2.82 0.34 Mn0.67Zr0.33Oy-反应后 33.91 54.57 11.51 4.74 0.32 Mn0.8Zr0.2Oy 51.27 40.37 8.36 4.83 0.33
下载: 导出CSV
-
[1] TANG X, MISZTAL P K, NAZAROFF W W, et al. Volatile organic compound emissions from humans indoors[J]. Environmental Science & Technology, 2016, 50: 12686-12694. [2] LIU F, CAO R, RONG S, et al. Tungsten doped manganese dioxide for efficient removal of gaseous formaldehyde at ambient temperatures[J]. Materials & Design, 2018, 149: 165-172. [3] BAI B, QIAO Q, ARANDIYAN H, et al. Three-dimensional ordered mesoporous MnO2-Supported Ag nanoparticles for ctalytic removal of formaldehyde[J]. Environmental Science & Technology, 2016, 50: 2635-2640. [4] YUSUF A, SNAPE C, HE J, et al. Advances on transition metal oxides catalysts for formaldehyde oxidation: A review[J]. Catalysis Reviews, 2017, 59: 189-233. doi: 10.1080/01614940.2017.1342476 [5] QI L, CHENG B, YU J, et al. High-surface area mesoporous Pt/TiO2 hollow chains for efficient formaldehyde decomposition at ambient temperature[J]. Journal of Hazardous Materials, 2016, 301: 522-530. doi: 10.1016/j.jhazmat.2015.09.026 [6] BOHM M, SALEM M Z, SRBA J. Formaldehyde emission monitoring from a variety of solid wood, plywood, blockboard and flooring products manufactured for building and furnishing materials[J]. Journal of Hazardous Materials, 2012, 221-222: 68-79. doi: 10.1016/j.jhazmat.2012.04.013 [7] WANG R, ZHU R, ZHANG D. Adsorption of formaldehyde molecule on the pristine and silicon-doped boron nitride nanotubes[J]. Chemical Physics Letters, 2008, 467: 131-135. doi: 10.1016/j.cplett.2008.11.002 [8] PEI J, ZHANG J S. On the performance and mechanisms of formaldehyde removal by chemi-sorbents[J]. Chemical Engineering Journal, 2011, 167: 59-66. doi: 10.1016/j.cej.2010.11.106 [9] ZHAO D-Z, LI X-S, SHI C, et al. Low-concentration formaldehyde removal from air using a cycled storage–discharge (CSD) plasma catalytic process[J]. Chemical Engineering Science, 2011, 66: 3922-3929. doi: 10.1016/j.ces.2011.05.019 [10] LIANG W J, LI J, LI J X, et al. Formaldehyde removal from gas streams by means of NaNO2 dielectric barrier discharge plasma[J]. Journal of Hazardous Materials, 2010, 175: 1090-1095. doi: 10.1016/j.jhazmat.2009.10.034 [11] KIBANOVA D, SLEIMAN M, CERVINI-SILVA J, et al. Adsorption and photocatalytic oxidation of formaldehyde on a clay-TiO2 composite[J]. Journal of Hazardous Materials, 2012, 211-212: 233-239. doi: 10.1016/j.jhazmat.2011.12.008 [12] NIE L, YU J, JARONIEC M, et al. Room-temperature catalytic oxidation of formaldehyde on catalysts[J]. Catalysis Science & Technology, 2016, 6: 3649-3669. [13] ZHANG C, HE H, TANAKA K-I. Catalytic performance and mechanism of a Pt/TiO2 catalyst for the oxidation of formaldehyde at room temperature[J]. Applied Catalysis B: Environmental, 2006, 65: 37-43. doi: 10.1016/j.apcatb.2005.12.010 [14] CHEN T, DOU H, LI X, et al. Tunnel structure effect of manganese oxides in complete oxidation of formaldehyde[J]. Microporous and Mesoporous Materials, 2009, 122: 270-274. doi: 10.1016/j.micromeso.2009.03.010 [15] TAN Y, ZHU F, WANG H, et al. Noble‐Metal‐Free metallic glass as a highly active and stable bifunctional electrocatalyst for water splitting[J]. Advanced Materials Interfaces, 2017, 4. [16] FARBER C, STEGNER P, ZENNECK U, et al. Teaming up main group metals with metallic iron to boost hydrogenation catalysis[J]. Nature Communications, 2022, 13: 3210. doi: 10.1038/s41467-022-30840-4 [17] MIAO L, WANG J, ZHANG P. Review on manganese dioxide for catalytic oxidation of airborne formaldehyde[J]. Applied Surface Science, 2019, 466: 441-453. doi: 10.1016/j.apsusc.2018.10.031 [18] TSONCHEVA T, IVANOVA L, PANEVA D, et al. Cobalt and iron oxide modified mesoporous zirconia: Preparation, characterization and catalytic behaviour in methanol conversion[J]. Microporous and Mesoporous Materials, 2009, 120: 389-396. doi: 10.1016/j.micromeso.2008.12.007 [19] COUZON N, BOIS L, FELLAH C, et al. Manganese oxidation states repartition in a channel-like mesoporous zirconium oxide[J]. Journal of Porous Materials, 2020, 27: 1823-1835. doi: 10.1007/s10934-020-00962-5 [20] DENG Z, WANG M, ZHANG H, et al. Mn–Zr composite oxides as efficient catalysts for catalytic oxidation of vinyl chloride[J]. New Journal of Chemistry, 2023, 47: 9212-9221. doi: 10.1039/D2NJ05964A [21] 焦坤灵, 焦晓云, 刘佳杰, 等. Mn/Zr改性稀土尾矿催化剂NH3-SCR脱硝机理分析[J]. 中国环境科学, 2023, 43: 5655-5662. doi: 10.3969/j.issn.1000-6923.2023.11.003 [22] 谈冠希, 迟姚玲, 李双, 等. 锰锆复合氧化物CO催化还原NO性能研究[J]. 燃料化学学报, 2019, 47: 1258-1264. [23] BULAVCHENKO O A, VINOKUROV Z S, AFONASENKO T N, et al. Reduction of mixed Mn–Zr oxides: in situ XPS and XRD studies[J]. Dalton Transactions, 2015, 44: 15499-15507. doi: 10.1039/C5DT01440A [24] OUYANG J, ZHAO Z, SUIB S L, et al. Degradation of Congo Red dye by a Fe2O3@CeO2-ZrO2/Palygorskite composite catalyst: Synergetic effects of Fe2O3[J]. Journal of Colloid and Interface Science, 2019, 539: 135-145. doi: 10.1016/j.jcis.2018.12.052 [25] YAN Q, LI X, ZHAO Q, et al. Shape-controlled fabrication of the porous Co3O4 nanoflower clusters for efficient catalytic oxidation of gaseous toluene[J]. Journal of Hazardous Materials, 2012, 209-210: 385-391. doi: 10.1016/j.jhazmat.2012.01.039 [26] TODOROVA S, NAYDENOV A, KOLEV H, et al. Mechanism of complete n-hexane oxidation on silica supported cobalt and manganese catalysts[J]. Applied Catalysis A: General, 2012, 413-414: 43-51. doi: 10.1016/j.apcata.2011.10.041 [27] DUAN C, MENG M, HUANG H, et al. Effect of calcination temperature on the structure and formaldehyde removal performance at room temperature of Cr/MnO2 catalysts[J]. Research on Chemical Intermediates, 2022, 48: 2705-2720. doi: 10.1007/s11164-022-04713-w [28] LIU P, HE H, WEI G, et al. Effect of Mn substitution on the promoted formaldehyde oxidation over spinel ferrite: Catalyst characterization, performance and reaction mechanism[J]. Applied Catalysis B: Environmental, 2016, 182: 476-484. doi: 10.1016/j.apcatb.2015.09.055 [29] LU S, ZHENG F, WANG H, et al. Engineering MnO2 nanotubes@Co3O4 polyhedron composite with cross-linked network structure for efficient catalytic oxidation of formaldehyde[J]. Catalysis Letters, 2023, 154: 2949-2962. [30] BAI B, LI J, HAO J. 1D-MnO2, 2D-MnO2 and 3D-MnO2 for low-temperature oxidation of ethanol[J]. Applied Catalysis B: Environmental, 2015, 164: 241-250. doi: 10.1016/j.apcatb.2014.08.044 [31] YE Q, ZHAO J, HUO F, et al. Nanosized Au supported on three-dimensionally ordered mesoporous β-MnO2: Highly active catalysts for the low-temperature oxidation of carbon monoxide, benzene, and toluene[J]. Microporous and Mesoporous Materials, 2013, 172: 20-29. doi: 10.1016/j.micromeso.2013.01.007 [32] WANG J, LI J, JIANG C, et al. The effect of manganese vacancy in birnessite-type MnO2 on room-temperature oxidation of formaldehyde in air[J]. Applied Catalysis B: Environmental, 2017, 204: 147-155. doi: 10.1016/j.apcatb.2016.11.036 [33] WANG J, ZHANG P, LI J, et al. Room-temperature oxidation of formaldehyde by layered manganese oxide: Effect of water[J]. Environmental Science & Technology, 2015, 49: 12372-12379. [34] CAI T, HUANG H, DENG W, et al. Catalytic combustion of 1, 2-dichlorobenzene at low temperature over Mn-modified Co3O4 catalysts[J]. Applied Catalysis B: Environmental, 2015, 166-167: 393-405. doi: 10.1016/j.apcatb.2014.10.047 [35] QIAN J, MO J, ZHOU Y, et al. Study of manganese–cerium composite oxide catalysed oxidation for low concentration formaldehyde at room temperature[J]. Materials Chemistry and Physics, 2022, 285: 126151-126167. doi: 10.1016/j.matchemphys.2022.126151 [36] 周辉, 步宇婷, 唐兢, 等. 温和条件下MnO2催化剂的制备及其降解甲醛的研究[J]. 现代化工, 2023, 43: 233-237. -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1012
- HTML全文浏览数: 1012
- PDF下载数: 10
- 施引文献: 0
